

EU Commission MDR/IVDR includes changes to medical device software classification, AI Act delay
BRUSSELS – The European Commission’s proposed revisions to the Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) include specific changes to the regulation of medical device software, including medical device artificial intelligence (AI), in Europe. According to experts who spoke at the 2026 DIA-RAPS Combination Products in the EU conference, the proposed changes would clarify how the products are classified and could delay the implementation of the EU Artificial Intelligence (AI) Act.Nada Alkhayat, a policy officer at the Commission, outlined the Commission’s approach to regulating medical device software (MDSW) and proposed legislative changes to implement it.
In December, the Commission proposed broad revisions to MDR and IVDR after years of challenges in implementing the regulations. In proposing the revisions, Alkhayat said the Commission also considered factors such as increased expectations of medical device software manufacturers to adopt higher standards and enhanced compliance. They also considered the impact of software classification rules, a heightened focus on post-market surveillance (PMS) and vigilance, the need to emphasize software risk management, and the need for software manufacturers to adopt a lifecycle approach.
Al Khayat said the Commission proposed changes to the regulation of medical device software to adopt a proportionate software classification approach. She noted that it has proposed targeted clarifications to Annex VIII of the MDR, including Rule 11, which outlines the classification rules for medical software.
She said the objective is to better align software classification with clinical risk, avoid up-classification of medical software when there is limited patient impact, and align regulations with the risk categorization developed by the International Medical Device Regulators Forum (IMDRF) for software as a medical device (SaMD), according to the table below:
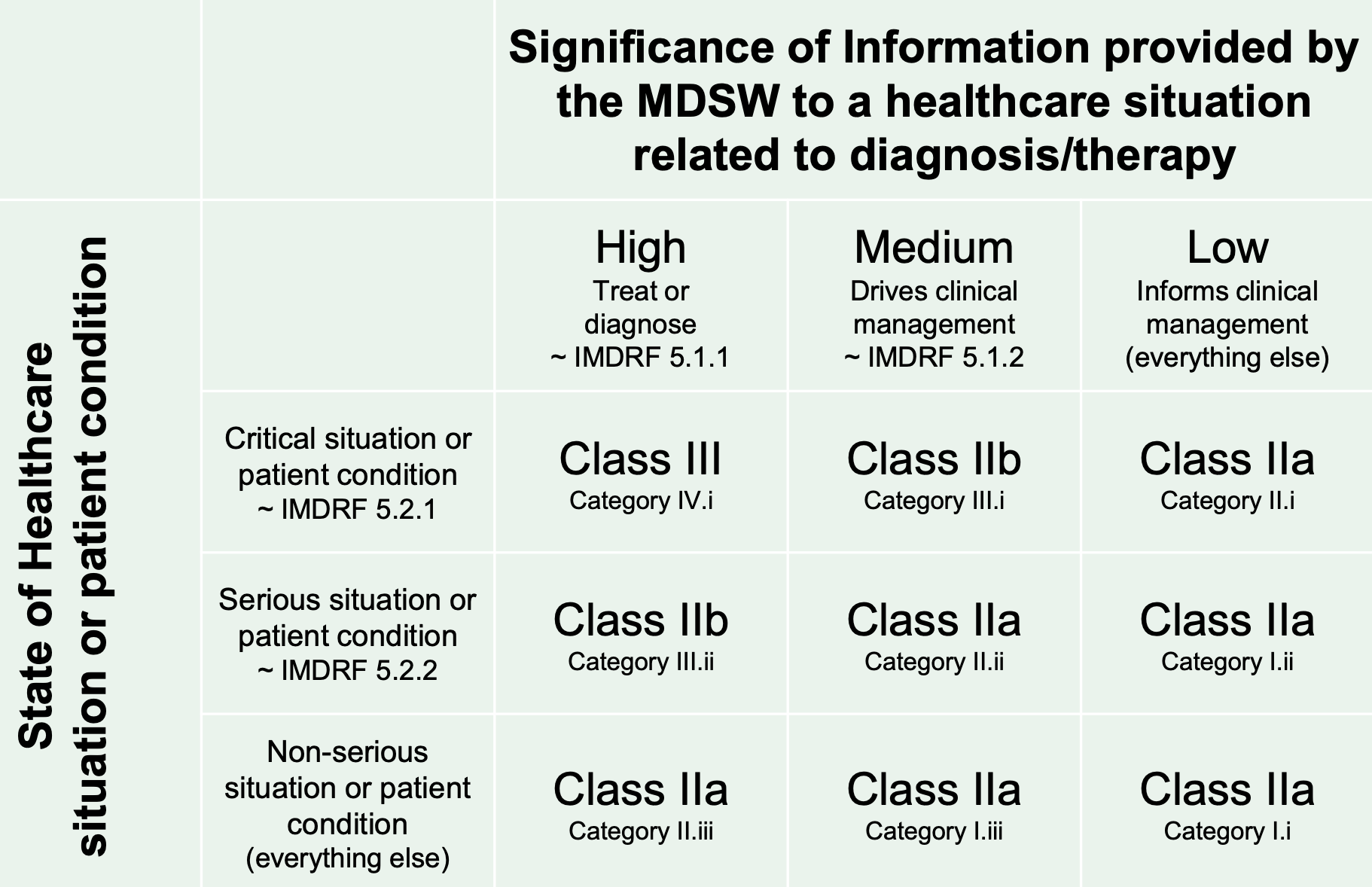
(Source: European Commission)
"It is very complicated to take a matrix and try to turn it into words, especially in legal terms," said Alkhayat. "We really tried our very best, but there is now the opportunity to have your say, in general, not just on this topic, to give your feedback if you think there needs to be some tweaks in order to better align it with what the aim is."
Alkhayat said the Commission has also proposed revisions to the MDR/IVDR that would add language on how the legislation interacts with the AI Act.
"With the AI act, there's a proposal for the clarification of rules and avoiding duplication, and essentially to ensure that the medical device framework remains the primary safety go-to framework for medical device AI, and reduce any possible duplication of activities or overlaps," she added.
Sebastian Fischer, a regulatory strategy principal at TÜV SÜD, also discussed the Commission's proposed revisions to MDR and IVDR that specifically apply to AI medical device products. He noted that the proposal would mean that MDR and IVDR would take precedence over the AI Act and AI Act requirements for medical device AI would effectively be moved into Annex I of the MDR and IVDR.
He also noted that the most important part of the proposal for medical device AI products is that it would extend the implementation of the AI Act by a year for high-risk devices under Article 6(1) of the act until August 2028.
"And now we are waiting for the omnibus to become adopted, which is hopefully the case before August this year," said Fischer. "There's slight concern that this might cause still some trouble, because member states are now obviously waiting for the central omnibus bill to [implement their national laws].
"Worst case could be that the one-year gain by the omnibus is entirely eaten up by member states now having to wait for it," he added.
In recent years, the US Food and Drug Administration has adopted predetermined change control plans (PCCPs) that allow medical device manufacturers to make product changes based on parameters agreed upon by regulators and manufacturers. That concept has also been adopted more recently by IMDRF in a PCCP guideline. (RELATED: IMDRF plans new PCCP guideline, adds new affiliates, Regulatory Focus 30 September 2025)
While PCCPs can apply broadly to medical devices, they have been used more frequently for software changes. Alkhayat notes that the Commission has proposed a lifecycle management approach that can use PCCPs to allow changes to software, including AI, and allow notified bodies to accept those product updates.
"We've integrated that in the commission proposal as well in order to facilitate changes for software and also in future AI [products], taking inspiration from the AI Act itself," she added.
Alkhayat said that another key proposal the Commission has made is the allowance of regulatory sandboxes, which enable regulators at the national and EU levels to test new regulatory ideas and principles. She also noted that regulators are facing more innovations and interplays between product areas that require an agile regulatory framework.
"If you don't have the tools to react when [innovation] comes at you, then we have to wait 15 more years in order to amend the regulations, in order to keep them with the state of the art," said Alkhayat. "It's really trying to find a proportionate way to respond to the growing innovation and technology in our field."

