

EU MDR survey: Time investment, costs, and personnel resources
In 2020, a survey was conducted among medical device manufacturers to assess the true costs of the EU Medical Device Regulation for companies. The results show that the regulation requires considerable resources on the part of manufacturers in terms of time, costs, and personnel. Almost half the respondents said they believed the regulation would cost more than 5% of their annual turnover. However, most still relied on manual processes for their clinical data capture activities, which may be adding to the burden.Introduction
Regulatory challenges for medical device manufacturers are increasing. With the transition period for the EU Medical Device Regulation (EU MDR 2017/745)1 ending in May 2021, medtech companies that produce or sell devices within the EU have to put the necessary processes in motion to comply with the new demands. In particular, the regulation places much greater emphasis on a lifecycle approach, as well as transparency. With regard to the lifecycle approach, once a device has been sold and is being used on the market, there has to be sufficient evidence available showing it is fulfilling EU safety and performance standards. For companies, this means gathering much more data on their devices than previously, and in many cases, gathering it more regularly as well.Unannounced audits by notified bodies will also become more frequent for certain device risk classes. It is not surprising, therefore, that most view the EU MDR as a costly and challenging matter. Yet, so far, there has been little data on the true monetary impact of the new regulation for medical device companies. For this reason, in 2020, we set out to ascertain how much time, money, and personnel resources companies believed the new regulation would require. We also wanted to find out how many of them had digitalized key processes, such as clinical data collection activities, in the postmarket phase. The purpose of this article is to summarize the main survey findings.2-4
Survey participants and methodology2
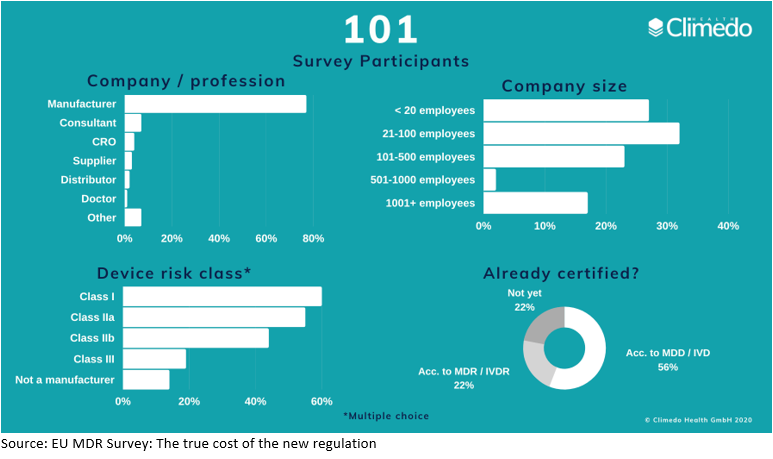
A total of 101 participants from Europe – most of them German-speaking – participated in the survey in August of 2020 (Figure 1). More than 75% of respondents worked for medical device manufacturers, and the remainder worked for distributors, suppliers, contract research organizations (CROs), or related consulting firms. To obtain a representative picture, we asked nonmanufacturers to answer all questions on behalf of their clients to the best of their knowledge. With regard to company size, 63% of respondents worked for organizations with fewer than 500 employees. This was to be expected, because most medtech companies in Europe are small- or medium-sized enterprises (SMEs).In regard to the medical devices participants produced or worked with, all device risk classes were represented, with almost 60% citing Class I; 57%, class IIa; 44%, Class IIb; and 21%, Class III. Most participants reported manufacturing a combination of different device classes. We also asked how many different product categories the companies worked with, and more than half (54%) worked with one to five product categories. Sixteen percent worked with 6 to 10 categories, and the remaining 30% worked with 11 or more product categories. Finally, the survey assessed whether the participants’ products were already EU MDR certified: 60% were still certified according to MDD (Medical Device Directive 93/42/EEC),5 only 21% were certified according to MDR, and 19% were not yet certified.
FIGURE 1 Survey participants
With regard to the survey methodology, we used our own eSurvey tool to gather the responses.
Time investments
In the first part of the survey, we wanted to find out how much time companies were investing in meeting the EU MDR demands overall, and which areas were proving most time consuming.Up to half a workday a week for the EU MDR
According to the responses, 55% of companies were investing more than 5 hours a week in fulfilling the MDR requirements; 35% invested between 1 and 5 hours, and 10% invested less than 1 hour a week. Therefore, for an employee with a 40-hour work week, this would mean investing more than half a working day a week, which is significant.
Time-consuming aspects of the EU MDR
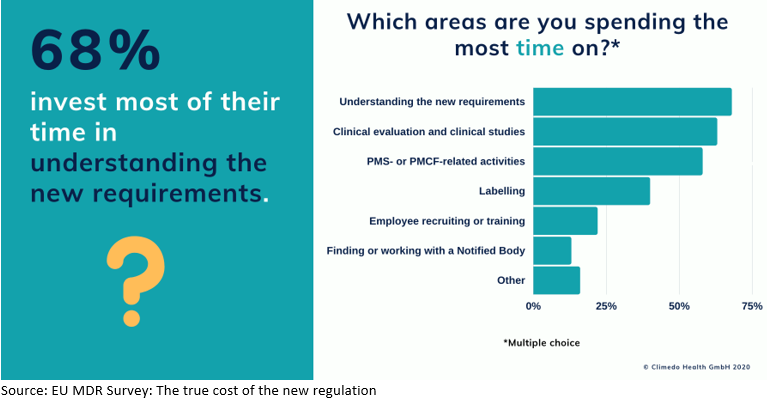
When participants were asked in a multiple-choice question on which specific areas they spent most of their MDR-related time, most – 68% – selected “Understanding the new requirements.” The EU MDR is a lengthy and complex document with many annexes and cross-references, so that response was not surprising. That was followed by 63% of respondents selecting “Clinical evaluation and clinical studies” as their most time-consuming aspect. Additional responses were “Postmarket surveillance (PMS) and Postmarket clinical follow-up (PMCF) activities” (58%), “Labelling” (40%), “Employee recruiting or training” (22%), “Finding or working with a notified body” (13%), and “Other” (16%; Figure 2).
FIGURE 2 Most time-consuming areas
Other time investments
In the “Other” field, respondents named “Gap analyses,” “Choosing harmonized standards,” “Setting up a quality management system,” “Creating contracts,” “Adapting existing documents, particularly risk management,” “Checking and archiving declarations of conformity,” and “Adapting technical files” as being particularly time consuming.
Financial investments
The following section focused on the financial implications of the new regulation.Costly aspects of the EU MDR
Analogous to the question about which areas required a lot of time, the next question’s purpose was to find out where most of the companies’ money was going.
It turned out that 75% of participants believed that clinical evaluations and clinical studies were the most expensive activity for their companies. As the previous question showed, this was also considered a highly time-consuming area. In second place, just over half of the respondents (52%) cited “PMS- or PMCF-related activities” as one of their most costly areas. With the EU MDR focusing more strongly on the lifecycle approach than the MDD, manufacturers are obliged to gather a significant amount of data in the postmarket phase to prove the safety and performance of their products once they are being applied by patients or medical staff.
Around 35% of respondents cited “Understanding the new requirements” as expensive, possibly because they had to hire consultants to help them make sense of the EU MDR demands. This was backed up by a handful of companies naming “Consultation services” in the open question field. “Labelling” and “Training and recruiting employees” were each considered as expensive by about 25% of respondents. Finally, just over 8% of respondents said that “Finding or working with a notified body” was expensive, which is interesting considering that there are still very few notified bodies certified under EU MDR. As of February 2021, there are fewer than 20, which is not enough to cover demand within the EU, and many of them are said to already be dealing with huge request backlogs.
Other costs
Responses in the “Other” field included “Consulting costs,” “Transferring data to EUDAMED (European Databank on Medical Devices),” “Checking the declaration of conformity,” “Validation modules,” and “Implementing the requirements.” One company also noted that the costs and labor resulting from the EU MDR are not manageable for SMEs.
Total cost for companies
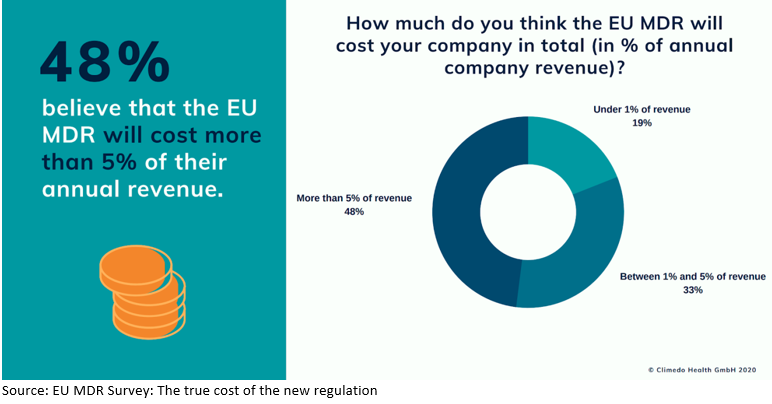
Regarding the overall costs, we wanted to know what the companies believed the new regulation would cost them as a proportion of their annual revenue (Figure 3). Here, almost half (48%) of respondents said they thought it would cost them more than 5% of their annual revenue, a third (33%) estimated that it would cost between 1% and 5%, while less than a fifth (19%) believed it would be less than 1%. Those responses suggest the EU MDR is a very costly matter for the majority of affected companies.
FIGURE 3 Total costs of the EU MDR in relation to annual revenue
Personnel resources
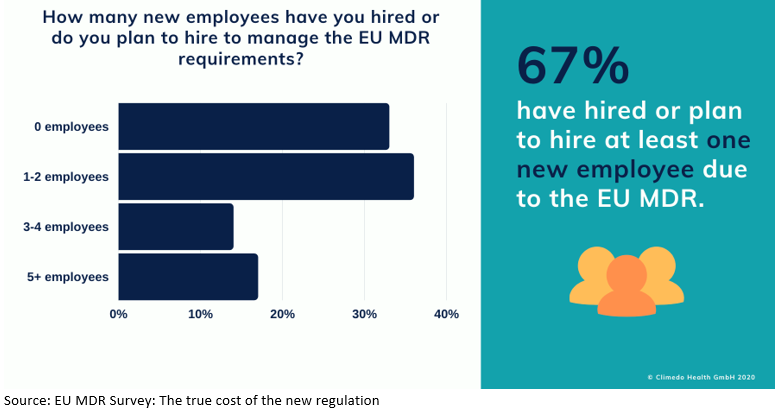
We have seen that dealing with the new regulation demands a great deal of time and money from companies, so we also wanted to find out about human resources. We asked participants whether they had hired, or were planning to hire, any new employees to tackle the EU MDR, and if so, how many.About a third (33%) said they had not hired, and were not planning to hire, any new staff. Just over 36%, however, planned to hire or had hired one to two new members of staff, while 14% were hiring three to four, and 17% were hiring more than 5 (Figure 4). Larger companies tended to hire more staff to meet their needs. As the responses to previous questions showed, manufacturers are also investing a great deal of resources in consulting services, as well as hiring their own personnel. Therefore, it would seem that EU MDR compliance also requires a great deal of “brainpower” in addition to time and money.
FIGURE 4 The need for more personnel
Clinical data needed for the EU MDR
In the next section, we wanted to assess how companies were capturing clinical data related to their medical devices, and how much time and effort this was costing them. Since PMCF is a key part of PMS, we decided to focus on this area.Systems used for PMCF-related data capture
Our first question in this segment aimed to assess which tools and technologies companies were applying to gather clinical data for their PMCF activities. In this multiple-choice question, more than two-thirds (69%) of the respondents said they were using Excel spreadsheets, while almost half (47%) said they were using paper (Figure 5). In addition, 12% claimed to be using a survey tool, since part of PMCF can also be conducted using surveys among end users, such as physicians, medical staff, or patients. Finally, just under 10% had implemented an electronic data capture tool, a computer-based system designed to capture clinical data in an electronic format.
FIGURE 5 Systems used for PMCF-related data capture
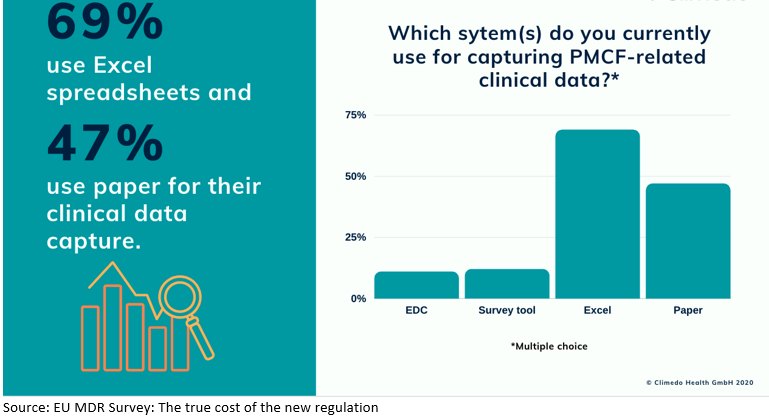
Manual methods, such as paper or Excel, are likely to struggle in the post-EU-MDR era, especially with the amount of data required and in the face of unannounced audits, for which data needs to be made accessible in a quick and straightforward manner.
Time spent on documentation and data management
Exploring more deeply, we asked how much time companies spent on administrative tasks, such as documentation and data management, as a percentage of the total time they spend on their clinical studies or PMCF activities. For 13% of companies, documentation and data management took up more than a quarter of their clinical study- or PMCF-related time. Forty-three percent said they spent between 15% and 25% of time on documentation and data management, and 27% spent between 10% and 15% on these activities.Onboarding new users
Getting new users up to speed with a clinical study or survey is crucial and can be elaborate, depending on the system used. We asked our survey participants how many hours they spent onboarding users, such as study coordinators or hospital staff. A small number of respondents (6%) said it took them more than 20 hours, which is about half a working week. Less than a fifth (17%) said they spent between 10 and 20 hours on onboarding, and just under a third (32%) said they required between 5 and 10 hours for this activity. Finally, almost half (45%), spend less than 5 hours on their onboarding activities, which is encouraging. Nevertheless, more than half still spend more than 5 hours on this task, which is not insignificant.
Communication with stakeholders
In the next question, we wanted to find out how many hours a week companies typically spent communicating with key stakeholders of their PMCF studies, such as doctors, CROs, or study coordinators. Almost a tenth (9%) of respondents said they spent more than 3 hours a week on this type of communication, while more than a third (36%) spent between 1 and 3 hours on it. More than half (55%) said they spent less than 1 hour a week on it. This suggests that stakeholder communication in PMCF is not a particularly time-consuming matter for most participants.
Automation of PMCF processes
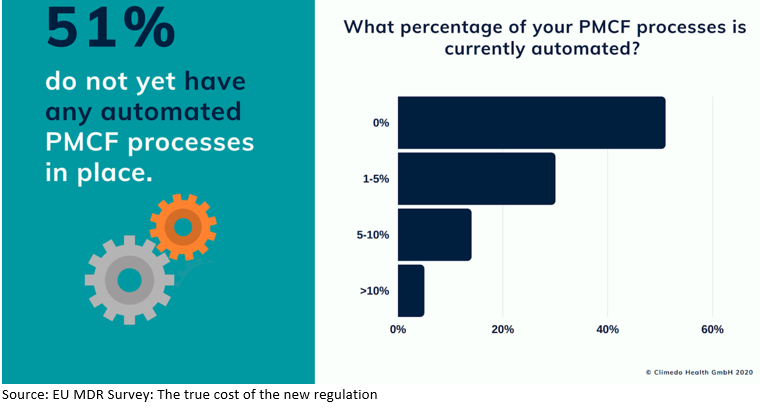
Finally, we assessed the level of automation of medtech companies’ PMCF processes, because automating a large part of these activities could save a lot of time for those conducting the studies and surveys. Just over half (51%), however, had not yet automated any part of their PMCF processes at the time of the survey (Figure 6). Almost a third (30%) had automated between 1% and 5% of these processes, whereas 13% had automated between 5% and 10%, and just 5% had automated a tenth of their processes. That suggests there is definitely still room for improvement from a digitalization point of view in this respect.
FIGURE 6 The level of automation for PMCF processes
Conclusion
The survey results show that managing the EU MDR demands is costing companies a great deal of time, money, and personnel resources. Moreover, the postponement of the regulation’s validity date to May 2021 did not seem to alleviate the situation.On the one hand, the EU Commission should create the necessary framework to ensure that the regulatory system is operational, that manufacturers can make a smooth transition to MDR, and that their bringing new products to market is not hindered further. On the other hand, there are areas in which manufacturers themselves can take action, for example, by digitalizing their clinical data capture, many processes can be automated, which in turn saves valuable resources. This typically results in far fewer errors compared with paper or Excel spreadsheets as well. Moreover, removing a large part of their administrative work can allow companies to focus more of their time and efforts on improving their medical devices and gaining a competitive advantage.
Despite its many challenges, the EU MDR will bring several benefits in the long term. Safety and performance standards will be scrutinized more closely throughout devices’ lifecycle, meaning that older, possibly unsafe devices will be removed from the market altogether. Furthermore, the amount of new data gathered will offer companies unprecedented access to insights which can help improve their devices, shifting from mere compliance to higher levels of quality and innovation. This can also result in new product claims for the sales and marketing teams. Finally, the EUDAMED database6 will create greater transparency for all stakeholders, enabling patients to make informed healthcare decisions about devices by researching them ahead of prescription or usage. Patients with implants will also receive a registration card with access to information about the manufacturer and their safety records.
If the EU MDR is managed in the right way by authorities and manufacturers, patients will be able enjoy a higher level of safety and transparency, while companies will be able to improve their products, reputation and – in the long run – their revenues. The new regulation should therefore not be viewed as a mere cost driver, but as a potential catalyst for long-term innovation.
References
- European Parliament and Council of the EU. Regulation (EU) 2017/745 of the European parliament and of the council of 5 April 2017 on medical devices. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745. Accessed 25 February 2021.
- Climedo Health. EU MDR Survey – The true cost of the new regulation. https://discover.climedo.com/en/eu-mdr-survey-results-true-cost-of-the-new-regulation. Conducted July-August 2020. Accessed 25 February 2021.
- Taylor NP. EU MDR costing smaller medtechs 5% of their annual sales: Survey. MedtechDive website. https://www.medtechdive.com/news/eu-mdr-costing-smaller-medtechs-5-of-their-annual-sales-survey/584399/. Published 31 August 2020. Accessed 25 February 2021.
- Medical Plastics News website. The true cost of EU MDR: Survey results released. https://www.medicalplasticsnews.com/news/the-true-cost-of-eu-mdr-survey-results-released/. Published 27 August 2020. Accessed 25 February.
- European Parliament and Council of the EU. Council Directive 93/42/EEC of 14 June 1993 concerning medical devices. https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:1993L0042:20071011:en:PDF . Accessed 25 February 2021.
- European Commission. Medical devices – EUDAMED. https://ec.europa.eu/health/md_eudamed/overview_en. Last updated February 2021. Accessed 25 February 2021.
About the authors
Veronika Schweighart, MS, is cofounder and COO of Climedo Health, where she oversees human resources, marketing, and sales, and works with clients on digital solutions for their clinical data management. She has a BA degree in global business studies and a master of science degree in business studies. Schweighart has 4 years’ experience in regulatory affairs and is a member of several European medtech clusters. She can be contacted at [email protected].
Catherine Higginson, MS, is marketing and communications manager at Climedo Health, where her duties encompass public relations, conducting market research around the medtech sector, organizing expert events on the EU MDR, and clinical data capture. Higginson has a BA Hons degree in modern languages and a master of science degree in management. She has 2 years’ experience in regulatory affairs and is a member of several European medtech networks. She can be contacted at [email protected].
Citation Schweighart V, Higginson C. What are the true costs of the EU MDR for companies? Regulatory Focus. February 2021. Regulatory Affairs Professionals Society.
©2021 Regulatory Affairs Professionals Society