

Regulatory Explainer: Understanding the Regulation of Generic Drug Labels
Regulatory Focus' ongoing series of Regulatory Explainers take complicated regulatory topics and make them simple enough for anyone to understand.
In our latest Regulatory Explainer, we are taking a look at a new attempt by the US Food and Drug Administration (FDA) to change the way it regulates generic drug labeling and the controversy that it is causing within industry.
Alright, Start From the Beginning: What's a Generic Drug?
In general, most drugs in the US are approved in one of two ways: Either as a new drug product (such as a new chemical entity never before approved, or at a never-before-approved dose) through a new drug application (NDA), or as an exact copy of an already-approved drug through the abbreviated new drug application (ANDA) pathway.
The ANDA pathway, otherwise known as the 505(j) pathway, allows for the approval of drugs that are generic-a term never actually used in the statute or regulations-versions of an already-approved drug.
Under 21 CFR 314, what we know as generic drugs are those that "are the same"-"identical in active ingredient(s), dosage form, strength, [and] route of administration"-as a drug that has been approved by FDA. (See 21 CFR 314.92)
In some cases, generic drugs can also be approved even if a drug's original application has been withdrawn, or may be declared the "same" (even if they have slight differences) by what's known as a suitability petition (see 21 CFR 314.93).
But this last example notwithstanding, the most important physical requirement for generic drugs is that they be shown to be bioequivalent (BE) to the reference-listed drug (RLD) (i.e. the originally approved product).
Bioequivalence refers to the "absence of a significant difference" between the bioavailability-specifically the extent and rate of absorption-of two (supposed) pharmaceutical drug equivalents over the course of a period of time, at the same dose and under the same conditions. "The generic version must deliver the same amount of active ingredients into a patient's bloodstream in the same amount of time as the pioneer drug," FDA explains on its website.
Drugs that are deemed to be bioequivalent are, for regulatory purposes, essentially the same.
Importantly, BE testing does not require a company to initiate lengthy or expensive clinical testing. The "abbreviated" way the application is approved is that it relies on the clinical testing of the original, already-approved drug. Instead of testing a drug on thousands of patients, it will likely only be tested on several dozen in tests to confirm bioequivalency.
By indicating to FDA that the drug is the "same," the product is therefore determined to have the same safety and effectiveness as the original drug.
How Else is a Generic Drug the Same as its Originator?
The most noticeable similarity is found in a generic drug's label, which must bear the exact same "conditions of use" statement as the original drug (unless the original drug has additional patents covering specific claims that have not yet expired).
But let's backtrack for a moment and talk about what a drug label is. Drug labeling is regulated by FDA under 21 CFR 201, and is defined as any information found on or associated with a drug product.
For example, it includes the text on the label affixed directly to the drug product, but also includes things like a package insert or prescribing information found in (i.e. with) or on the drug carton (often a box containing the drug bottle, which contains the drug).
Labeling is important because it informs healthcare professionals and consumers about what a drug is intended for (efficacy) and what its limitations are, including known adverse events. Under 21 CFR 201.56-7, drug labels must contain statements explaining a drug's known adverse reactions, major safety risks (i.e. Boxed Warnings), potential drug interactions, general warnings, contraindications, and recommended dose and routes of administration.
While these requirements apply to innovative pharmaceutical products, they also apply to generic pharmaceuticals approved under the ANDA pathway. Federal regulations at 21 CFR 314.92 require generic drugs to bear the same "conditions of use" statement as the original drug.
21 CFR 314.94(a)(8)(iv) goes even further, requiring that ANDA applicants have labeling that is "the same as the labeling approved for the reference listed drug." (Notwithstanding method of use patents or manufacturer information.)
In short: Generic drugs, in addition to being bioequivalent, must also bear the same labeling statements with regard to safety and efficacy.
Ok, So Generic Drugs Are Supposed to Have the Same Label. Why Would FDA Propose Changes?
Let's talk about fun stuff for a moment. Specifically, generic drug law.
The modern generic drug pathway was created in 1984 under the Drug Price Competition and Patent-Term Restoration Act, more commonly known just as the Hatch-Waxman Act. While generic drugs could be approved prior to 1984 under various pathways (the Drug Efficacy Study Implementation, or DESI program; as "paper NDAs" starting in 1978; or under a monograph), 1984 would mark the creation of a formal generic drug approval pathway. (1)
Lawsuits against drug companies, though, have been around for a long time. Many lawsuits against drug companies allege a "failure to warn"-that is, a failure to warn consumers about drug risks that were known to the company.
For example, if a company manufactured a cholesterol-lowering drug that killed some patients by causing heart attacks, and the company was found to have known about those risks but failed to warn consumers, it might then be sued by a consumer.
But for generic drugs, this posed a tricky legal question.
Assume for a moment that a generic company learned about a rare adverse event associated with its drug, one never before discovered by the original manufacturer of the drug. Even if it wanted to update the label of its drug, it would not be allowed to.
That's because the generic drug label needs to be the same as the original drug's labeling. If the generic drug were to add new risks to its label, it would no longer meet the sameness requirements under 21 CFR 314.
Are Generic Drug Companies Typically Found Guilty in Failure to Warn Cases?
No. In fact, they're nearly immune from them based on three recent Supreme Court decisions: Mutual v. Bartlett (2013), Pliva v. Mensing (2011) and Wyeth v. Levine.
In Levine, the court ruled that FDA approval does not prevent plaintiffs from suing manufacturers under state laws using so-called "failure to warn" theories of legal liability.
In Mensing, a patient who developed tardive dyskinesia after taking a generic drug, metoclopramide (generic Reglan) sued the manufacturer of the drug. However, the Supreme Court ruled that the generic manufacturers were not liable under Vermont tort law. In short: While generic pharmaceutical companies were required to report adverse events to FDA-as are all pharmaceutical companies-they aren't able to be sued by third parties in state courts.
The Supreme Court found in Mensing that generic companies are responsible to communicate any adverse events discovered to FDA (a long-standing obligation required of all pharmaceutical companies), but that generic companies are not legally liable to third parties (i.e. injured plaintiffs) in state courts.
Accordingly, Mensing modified Levine to allow failure-to-warn suits against NDA holders, but not ANDA holders.
In the Bartlett decision, another injured plaintiff had taken a generic NSAID, sulindac, to treat pain in her shoulder. The plaintiff developed toxic epidermal necrolysis, severely and permanently disfiguring her and leaving her nearly blind.
The crux of the case: Is a generic drug "defective" if it cannot adequately warn consumers about potential side effects?
The Supreme Court ruled that the generic drugs were not defective, and further that FDA's labeling update process preempted state laws, creating an even stronger barrier to future "failure to warn" lawsuits.
How are Drug Labels Updated?
Once a drug is approved, its labeling can be updated in a number of ways, as explained in 21 CFR 314.70.
- If a drug is seeking to update its label to reflect a new indication for use, it needs to submit a New Drug Application (NDA) referencing clinical trials data.
- A Prior Approval Supplement (PAS) may be required to be submitted if FDA requests or required that a drug label be updated in light of new safety developments, or if the manufacturer (including ANDA holders) proposes other substantial changes to the label.
- Annual Reports may be used to reflect very minor changes, such as an extension of an expiration dating period for a product.
A fourth category of updates is known as a Changes Being Effected (CBE) supplement. CBE notices come in two varieties:
- CBE-30: Changes must be submitted to FDA at least 30 days prior to a change going into effect to give FDA the chance to review it.
- CBE-0: Changes may be initiated immediately, but FDA must be notified immediately as well.
CBEs are meant to allow manufacturers to make "moderate changes" (21 CFR 314.70(c) and (c)(6)) to applications. In particular, CBE-0s are often used to make additions to drug labels to reflect new safety information.
Under 21 CFR 314.70(c)(6)(iii), FDA states that changes may be made to the labeling by the CBE process:
A.) To add or strengthen a contraindication, warning, precaution, or adverse reaction for which the evidence of a causal association satisfies the standard for inclusion in the labeling under 201.57(c) of this chapter;
B.) To add or strengthen a statement about drug abuse, dependence, psychological effect, or overdosage;
C.) To add or strengthen an instruction about dosage and administration that is intended to increase the safe use of the drug product;
D.) To delete false, misleading, or unsupported indications for use or claims for effectiveness; or
E.) Any labeling change normally requiring a supplement submission and approval prior to distribution of the drug product that FDA specifically requests be submitted under this provision.
FDA also allows ANDA holders to use the CBE-0 process to update its labeling to conform with changes made to the RLD's labeling or respond to specific FDA requests.
Why Might a Company Want to Update a Drug Label?
Under 21 CFR 314.80, 21 CFR 314.98, and 21 CFR 600.80, companies are required to keep track of adverse events that occur after a drug has been approved, regardless of whether their product was approved as a new drug or generic drug.
Because clinical testing on drugs prior to approval rarely (if ever) is able to determine the full range of side effects, this requirement ensures that companies and regulators can keep track of side effects that are rare, particularly dangerous, or only occur over a lengthy period of time (such as liver damage).
And under 21 CFR 201.57(c)(6)(i), companies are required to update their drug labeling as soon as "there is reasonable evidence of a causal association with a drug; a causal relationship need not have been definitely established."
Who can Update a Drug Label?
Only the holder of a new drug application (NDA) or biologics license application (BLA) (used to approve protein-based drug products) is permitted to update the label of a drug product.
To understand why, think back to the "sameness" requirements we referenced earlier at 21 CFR 314.94(a)(8)(iv). If the sponsor of a generic drug updated the label of its product to reflect new safety risks, it would no longer be "the same" as the RLD, thereby making it noncompliant with FDA regulations.
The need for this regulatory requirement has been contested, but branded manufacturers have raised a number of different reasons why adverse events should be consistent between the branded and generic versions of a drug. Two are of particular note:
- Sometimes generic drugs elicit different therapeutic responses owing to small differences between the RLD and the generic formulation. For example, in early 2013 FDA forced several generic versions of the antidepressant Wellbutrin XL off the market due to bioequivalence concerns.
- Sometimes the generic drug is not manufactured to the same standards of quality as the original drug. For example, Indian generic drug manufacturer Ranbaxy pleaded guilty in 2013 to having falsified data used to support approval of some of its generic drug products.
To manufacturers of branded drugs, then, the fact that a generic drug manufactured by a competitor has experienced a problem doesn't mean the RLD should be associated with the problem as well, especially as that might raise scrutiny of the drug.
In 1992, when FDA promulgated its regulations on generic drug labeling "sameness," the agency had a distinct reason for wanting drug labels to be the same. As it recounted in a 2013 Federal Register notice:
At the time of FDA's adoption of the generic drug regulations in 1992, FDA believed it was important that product labeling for the RLD and any generic drugs be the same to assure physicians and patients that generic drugs were, indeed, equivalent to their RLD.
However, it has recently adopted the stance that this approach is no longer needed. More on this later in the article.
If a Generic Drug Manufacturer is Aware of an Issue, How Can It Add that to the Label?
Labeling additions depend on specific circumstances, most notably including the severity of the adverse event.
- One is that the generic drug company approaches the owner of the RLD, which agrees that the adverse event is troubling, and the event gets added to the label through either a CBE or a PAS.
- Another is that the generic drug company makes FDA aware of the problem through a PAS, which requests that all companies manufacturing the drug update their labels to reflect the changes through the submission of a CBE/PAS. (See FDA's July 2013 Guidance on Safety Labeling Changes)
- Still another is that the generic drug company approaches the owner of the RLD, which does not agree that the label should be updated to reflect the adverse event.
- In still other cases, the NDA or BLA holder may no longer exist (such as if it declared bankruptcy or no longer markets the drug), meaning the generic drug manufacturer has no way of easily updating the label to reflect new safety risks.
If a generic drug manufacturer were to go ahead and update its label unilaterally, FDA would (in some cases) have the authority under 21 CFR 314.150(b)(10) to remove it from the market.
What Happens When a Branded Drug Manufacturer Updates its Label? Must a Generic Company Stop Marketing its Product?
In theory, yes. In practice, no.
Current FDA practice, as reflected in the May 2000 guidance document Revising ANDA Labeling Following Revision of the RLD Labeling, allows for amounts of time when the label of the generic drug is different from the RLD after the latter has been updated.
As FDA writes in the guidance: "OGD believes that prompt revision, submission to the Agency, and implementation of revised labeling are important to ensure the continued safe and effective use of generic drug products. Because the regulations state that the labeling of the generic must be the same as the innovator, the revision should be made at the very earliest time possible."
While FDA doesn't explain in the guidance what legal authority it has to allow this departure from the sameness requirement, in practice it means that generic drug products can continue to be marketed throughout the labeling update practice. Otherwise, each time the label of an RLD was updated, the generic would need to stop labeling the product while its labeling update application was approved by FDA.
This has not been a theoretical concern. In 2010, the passage of the Patient Protection and Affordable Care Act (PPACA) contained provisions intended to allow FDA to approve an ANDA even if its RLD had undergone labeling changes within the last 60 days. The provisions followed attempts by RLD holders to update their labels just prior to approval, requiring FDA to delay approval until the generic drug company brought its labeling into harmony with the RLD.
OK, It's a Complex System. But is it Broken?
FDA has argued that yes, it is broken. After the Mensing and Bartlett decisions, FDA reevaluated its policies and determined that the way generic drug labels are updated is no longer adequate.
It bases this finding on several different factors and points:
- Incentives: In the wake of the Mensing decision, FDA said some generic drug companies had lost the incentive to keep their labels updated, and in particular those marketing generic versions of RLDs that had since been withdrawn or discontinued.
As FDA explains in the federal register, "As a result of the decisions in Wyeth v. Levine and Pliva v. Mensing, an individual can bring a product liability action for failure to warn against an NDA holder, but generally not an ANDA holder, and thus access to the courts is dependent on whether an individual is dispensed a brand name or generic drug. The Mensing decision alters the incentives for generic drug manufacturers to comply with current requirements to conduct robust postmarketing surveillance, evaluation, and reporting, and to ensure that the labeling for their drugs is accurate and up-to-date."
And since nearly 80% of prescriptions filled are for generic drugs, that leaves a significant amount of the population without legal recourse in the event of a failure-to-warn event. - A Changing Industry: As we explained earlier, when FDA promulgated its regulations on generic drug labeling "sameness" in 1992, it had a distinct reason for wanting drug labels to be the same. As it recounted in a 2013 Federal Register notice:
"At the time of FDA's adoption of the generic drug regulations in 1992, FDA believed it was important that product labeling for the RLD and any generic drugs be the same to assure physicians and patients that generic drugs were, indeed, equivalent to their RLD."
Now, more than two decades later, generic drugs command more than 80% of the market and are well-known to consumers and physicians alike. "FDA believes it is time to provide ANDA holders with the means to update product labeling to reflect data obtained through postmarketing surveillance, even though this will result in temporary labeling differences among products," it wrote. - Abandoned Products: As we briefly mentioned under the "Incentives" section, some generic drug products have remained on the market even as the RLD has either been withdrawn or is no longer marketed. Because these products are still legally required to be the same as the RLD, their labels are unable to be updated unless FDA orders them to be updated (which is rarely done due to resource constraints).
Accordingly, the actual risks associated with these products may be unknown to consumers, especially now that Mensing has removed the requirement for companies to keep labels up to date. While FDA has on occasion selected ANDAs to be the new "reference standard" (i.e. new RLD) in cases where the original NDA has been withdrawn, in practice this rarely happens. - Nature of Adverse Events: Studies have determined that most labeling changes take place 11 years after a drug has been approved-a time when a drug is likely to have generic competition. For the most serious changes, such as new boxed warnings and contraindications, the median is 10-13 years, depending on the exact change. This data underscores the "importance of persistent and vigilant postmarket drug safety surveillance," FDA says.
Since these events are happening at a time when generic competition is coming into effect, FDA's thinking goes, it should be easier for generic drug manufacturers to initiate those changes as well since they're likely to discover those problems.
Has FDA Proposed Any Changes to the Way Generic Drug Labels are Regulated?
It has. In November 2013, it unveiled a new proposed rule, Supplemental Applications Proposing Labeling Changes for Approved Drugs and Biological Products.
The rule would permit a sponsor of a generic drug to immediately issue a labeling change using a CBE-0 application for any safety-related change, starting a process intended to allow differences to exist between the generic drug, the RLD and other approved ANDAs on a "temporary basis."
The CBE would need to contain information such as the basis for the labeling change and available data to support the change, such as adverse event data, published literature or epidemiologic studies.
How Would This Proposed Labeling Update Process Work?
Upon the submission of a CBE-0 to FDA, an ANDA holder would immediately be permitted to distribute the drug "accompanied by the revised labeling." The CBE-0 submission would need to contain a copy of the revised labeling in what is known as Structured Product Label (SPL) format, allowing FDA to quickly post the proposed labeling to its website and the National Library of Medicine's DailyMed database.
ANDA holders would also be required to send the holder of the RLD both the labeling change and a copy of the information supporting the change. This requirement is waived if the original NDA has been withdrawn, but would otherwise "ensure that the NDA holder for the RLD is promptly advised of the newly acquired information that was considered to warrant the labeling change."
Other ANDA holders that manufacture the same generic drug would not be required to be notified, but would instead need to subscribe to a new FDA web page that will be created and dedicated to posting information regarding the CBE labeling supplements. Once the CBE labeling update process is known to them, they may then submit their own comments to FDA regarding the labeling change.
After the RLD holder is made aware of the CBE-0 supplement, it would need to submit its own CBE-0 supplement in the event that it disagreed with the proposed update, as any changes would affect both the generic and RLD owners' labeling, FDA said.
Both sets of CBEs (RLD and ANDA holders') would then be posted to FDA's website. FDA would then evaluate the CBEs as either meeting or failing to meet its CBE labeling criteria. Approval would take one of three forms:
- CBE Approved: The ANDA holder's CBE is approved without changes
- CBE Approved With Changes: FDA approves the CBE, but with changes
- CBE Not Approved: FDA issues a complete response indicating that there is no basis for a change to the CBE
If approved, all other ANDA holders would then have 30 days in which to submit their own CBE-0 supplements to bring their labeling into conformity with the approved changes. FDA said that if the NDA holder does not submit a supplement seeking approval by its own accord, FDA may require it to do so (See: Safety Labeling Changes), even if the drug has been withdrawn from the market.
FDA has posted a helpful diagram of this labeling update process in the Federal Register.
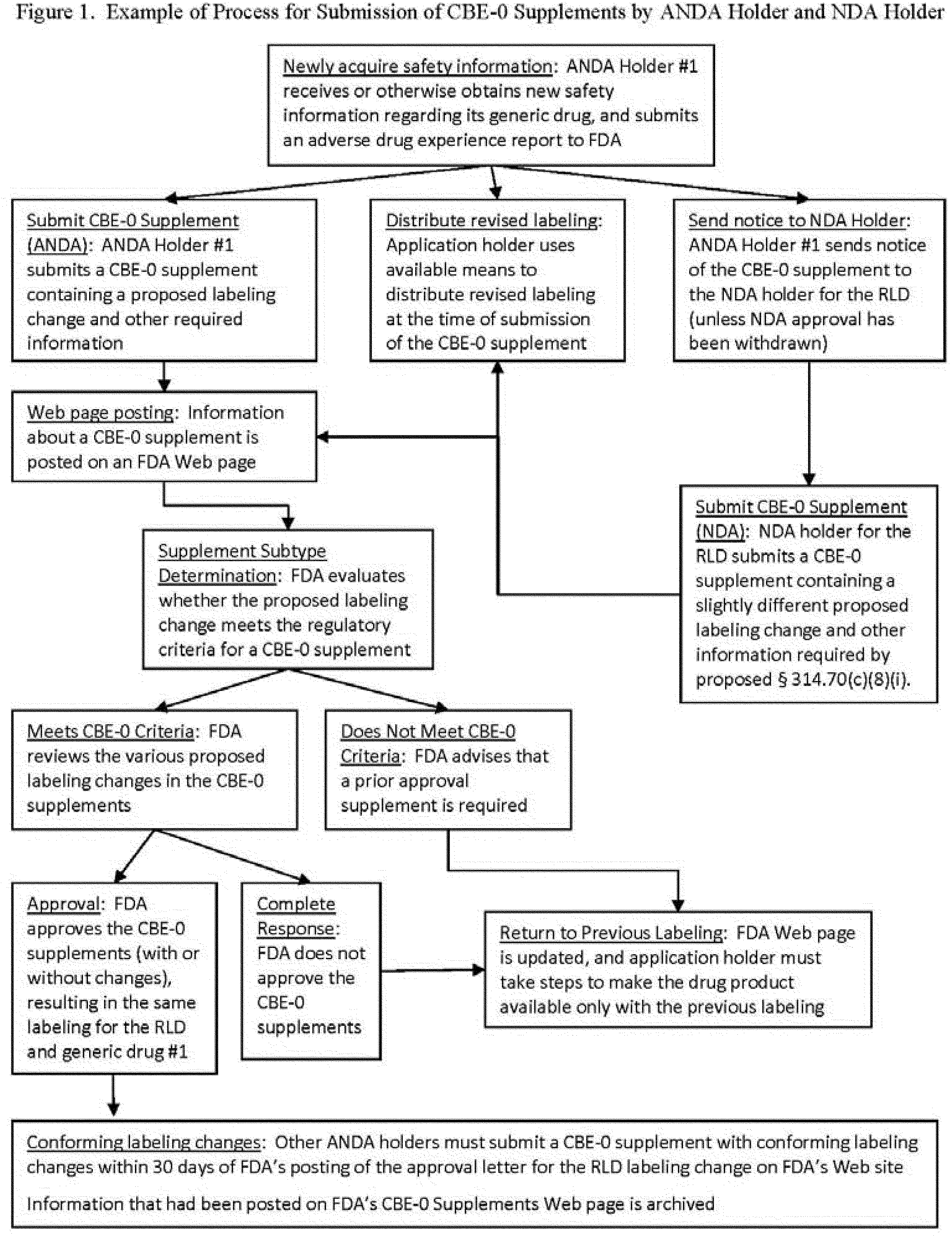{kind=link}
If a CBE is rejected by FDA, it must immediately "cease distribution of the drug product(s) accompanied by the revised labeling," and make the drug product "available only with the previous version of the label." FDA will withdraw generic drugs if the labeling "is no longer consistent with the labeling for the RLD, subject to certain exceptions." Companies would need to attach the previous package insert to the drug product "as soon as feasible," FDA said. FDA's proposed rule indicates that "time of next printing of product labeling for packaging" may be acceptable in such cases.
Anything Else I Need to Know About the Rule Itself?
In a departure, the proposed rule would also permit ANDA holders to distribute "Dear Health Care Provider Letters," which were previously only able to be sent by NDA holders. Those letters are intended to alert healthcare providers to major changes to a label.
Additionally, both ANDA and NDA holders would be able to use the CBE process to update the "Highlights" section of the drug labeling, which currently requires the submission of a prior approval supplement (PAS). FDA said current experience has found this limitation to be "unnecessary in practice."
How Has the Pharmaceutical Industry Reacted to FDA's Proposed Labeling Rule?
The industry, led by the Generic Pharmaceutical Association (GPhA), has responded forcefully against the proposal.
Industry's arguments track a number of varied approaches, which we'll try to summarize:
- Legal Concerns: Some legal experts have questioned if the rule is even legal. In its rule, FDA cites authority under 21 USC 301 and 42 USC 201, both of which allow it to declare a drug misbranded if it bears inadequate or insufficient warnings. It also claims authority under 21 USC 701 to regulate CBE supplements and their use. However, several legal experts say FDA's attempt to implement new rules conflicts with the "sameness" provisions of the Hatch Waxman Act.
"The law prohibits revisions that seek to change 'warnings' - which is precisely what the CBE provisions involved in the current proposal purport to do," wrote James Beck of the law firm Reed Smith. "Several maxims of statutory construction would seem to preclude the FDA from doing what it seeks to do here." (More Here and Here and Here) - Potential Confusion: FDA and industry alike have expressed concern that the update process could result in inconsistent drug labels across RLDs and their generic equivalents. This problem gets more complex if there are more generic competitors, as each ANDA holder is responsible for its own label and CBE process. For example, if there are 10 competitors to a popular blockbuster drug, there may be 11 labels, each of them with their own unique safety concerns.
- Liability: GPhA and the US Chamber of Commerce have both raised the concern that the rule would create new liability standards for generic drug manufacturers under the same failure-to-warn standards that have resulted in huge fines for many branded drug manufacturers. If generic manufacturers are required to implement the new rules, they may need to raise costs to deal with eventual settlements, or may decide to discontinue drugs with riskier profiles, thereby raising costs or causing drug shortages. According to a research report commissioned by GPhA, the rule could cost the industry an additional $4 billion per year.
- Physician Fatigue: FDA puts out a lot of communications each year-recall notices, safety updates, warnings, and more. So many, in fact, that some healthcare providers are simply unable to keep up with all of them and begin to ignore them. Some within industry have expressed concern that if generic manufacturers are now updating drug labels and sending "Dear Healthcare Provider" letters, doctors may begin to ignore the letters altogether, leading to worse levels of care for patients.
- New Legal Questions: Generic companies have expressed their concern that the current generic pharmaceutical legal environment, which has largely been made stable after 30 years of state and federal court decisions, could be upended. Of particular concern: What happens if a generic drug company petitions FDA to make a change, but the agency refuses? Could the company still be held liable for failing to warn consumers?
"It took 30 years to get to the Mensing decision - a clarification consistent with the Hatch-Waxman Statute and the regulatory framework that was 'hatched' from a delicate balance of the rights and responsibilities of the brand and generic industry in turn for certain period of patent and market exclusivity," Bob Pollack, former head of FDA's Office of Generic Drugs, wrote. "Let's make certain we don't create a bigger problem when trying to solve one that might have a more simplistic solution." - Lawsuits Against Branded Industry: Failure to warn lawsuits are now the exclusive purview of innovative companies with RLDs, and those companies have expressed concern that the issues raised by ANDA holders in CBE filings could increase their exposure to lawsuits. The theory is that any CBE filing could be a potential lawsuit-even ones that aren't added to the label.
- Other Options Available: A number of entities, and in particular GPhA, have proposed that the FDA itself take charge of the labeling process. "Since the FDA is the only entity with access to all the information and the expertise to evaluate and address this information, it is the only body in a position to decide whether a labeling change is warranted," wrote GPhA CEO Ralph Neas in a March 2014 op-ed arguing for FDA to be the sole arbiter and approver of labels. Labeling updates should be developed based on adverse event data and other information submitted by all companies-not just one or a few generic drug companies-he added.
PhRMA, the pharmaceutical industry's trade group, also called for a central role for FDA in approving labeling in its submitted comments. "Requiring FDA prior approval for all safety-related labeling changes in a multisource environment will help prevent potential confusion that might result from different versions of the same medicine having different labeling," the group wrote. - Unwarranted Concerns: At least one group, the Biotechnology Industry Organization (BIO), has expressed concern that the immediate posting of CBE submissions on FDA's website may raise safety concerns that end up being unwarranted. It has expressed similar concerns about ANDA holders' ability to send "Dear Healthcare Provider" letters.
- Input Into the Process: GPhA and Republican legislators have expressed concern that FDA consulted only one group during the formulation of its rule: a trade association representing trial attorneys. They have continued to press this issue, and in April 2014 demanded documents related to the meeting from FDA. (2)
How Have FDA and Other Groups Responded to These Concerns?
FDA and public advocacy groups like Public Citizen have been active in responding to concerns raised by industry groups, downplaying many of them.
A summary of those arguments may be found below:
- Increase in Liability Negligible: Groups argue that because generic drug companies were only recently given a waiver against liability, the increased liability concerns the companies reference in studies are dramatically overplayed. They argue that since the liability costs did not exist in the past, there is no reason for them to suddenly exist in the future, either.
- Legal Authority: FDA has continued to maintain that it does have the legal authority to implement its proposed rule. Janet Woodcock's testimony at a April 2014 legislative hearing provides a good overview of its legal argument, as does the proposed rule itself.
- Labeling Differences Already Exist: Recall what we wrote earlier when explaining a May 2000 guidance document, Revising ANDA Labeling Following Revision of the RLD Labeling. That guidance (which isn't legally binding) permits generic drugs to remain on the market after the RLD has updated its label so long as the ANDA label is updated "At the very earliest time possible." Since these differences already exist, critics say, there's little cause for alarm.
- Speed is the Key: FDA has deflected calls by GPhA, BIO and some legislators that FDA should use the PAS process instead of the CBE process to update labels. "The need to promptly communicate certain safety-related labeling changes based on newly-acquired information" reigns supreme, FDA said. The PAS process, while useful, "results in a delay in updating generic drug labeling and getting new information to prescribers and patients," FDA said in a response to congressional concerns.
- The Over-Warning Concerns are Overrated: The American Association for Justice, a trial lawyer advocacy group, has argued in comments that ANDA holders would not unnecessarily warn about potential risks associated with their products. "Generic drug manufacturers, like NDA holders, have significant financial incentives not to add unnecessary warnings to their labels," the group said in a filing with FDA. "If the risks of a drug are overstated on its label, doctors will be inclined to substitute other drugs indicated to treat the same medical conditions with fewer acknowledged dangers." The AAJ added that states generally only recognize "unreasonable failures to warn," and not ones where warnings were not justified by scientific evidence. FDA has supported this argument as well, saying that its CBE process will (or should) only be used for valid safety concerns.
- Vigilance: State attorneys general have argued that the rule will prompt generic pharmaceutical companies to be especially vigilant in tracking adverse events. While the law and FDA now require them to do so, the costs of noncompliance are considerably lower after the Mensing decision, they argued in a filing with FDA.
What Happens Next?
The comment period on the proposed rule, after being extended by two months, ended on 13 March 2014. The agency will now consider the comments received on the rule-there are many-and decide if the rule is worth moving forward either in its current form or with revisions.
If it does decide to move forward, FDA may release a final proposed rule by the end of the year, or likely by early 2015 at latest.
There are also growing indications that alternative models may be proposed, including by legislators. (2)
What Happens if the Rule Does go Into Effect?
Expect lawsuits from generic pharmaceutical manufacturers challenging the legality of the rule based on the "sameness" requirements of the Hatch-Waxman Act.
Where Can I Learn More About FDA's Proposed Generic Drug Labeling Rule?
- Proposed Rule
- OMB Information
- FDA Economic Impact Analysis
- Comments on Proposed Rule
- Proposed Rule Docket
Updates
- (1) - Updated on 10 April 2014 to reflect additional information about "generic" drug approvals prior to 1984.
- (2) - Updated on 23 April 2014 to reflect new concerns raised by GOP legislators, and new those legislators raising the prospect of "alternatives" to FDA's proposed rule.