

Global regulations governing orphan drug designation
Abstract Rare disease definitions and orphan drug designation (ODD) requirements vary across jurisdictions, shaping development strategy and evidence needs. This article examines orphan drug designation frameworks in the US, EU, UK, Japan, and China, highlighting differences in eligibility criteria, timelines, and incentives. It discusses how these differences affect evidence generation, sequencing of submissions, and long-term global planning for orphan medicines.
Keywords – orphan drug designation, rare disease definition, regulatory policy
Introduction
Rare disease definitions and ODD frameworks vary across regulatory jurisdictions, reflecting differences in regulatory philosophy, evidentiary standards, and health system context. Although a recent initiative by a multistakeholder panel of experts has developed a consensus on an operational definition of a rare disease,1 no universally harmonized global definition exists, and regional frameworks remain the primary basis for regulatory classification and policy implementation.2
ODD is the principal instrument used to support therapeutic development for rare diseases. While ODD frameworks share a common objective across regions, they differ substantially in eligibility criteria, evidentiary expectations at the designation stage, and the structure and timing of regulatory incentives.3-7 These differences extend beyond numerical prevalence thresholds and include jurisdiction-specific regulatory constructs, such as acceptance of orphan subsets, requirements to demonstrate comparative benefit, feasibility assessments, and reliance on formal disease catalogs. Such divergences have important implications for orphan drug development, influencing designation feasibility, early evidence generation, and regulatory engagement strategies across regions.
This article examines rare disease definitions and orphan drug designation frameworks in the US, EU, UK, Japan, and China, and how differing regulatory constructs shape global orphan drug development strategies. The article also explores how jurisdiction-specific ODD frameworks influence development feasibility and strategic decision making. By situating regulatory definitions within their broader health-system context, this article seeks to provide a practical understanding of how regulatory design affects downstream patient access.
Definitions and regulatory classification
Regulatory definitions of rare diseases underpin orphan drug designation frameworks and determine eligibility for regulatory incentives. Despite having shared policy objectives, regulatory authorities apply distinct conceptual criteria to define rarity, including prevalence thresholds, diagnostic requirements, and regulatory constructs such as orphan subsets, feasibility expectations, and catalog-based eligibility (Table 1).
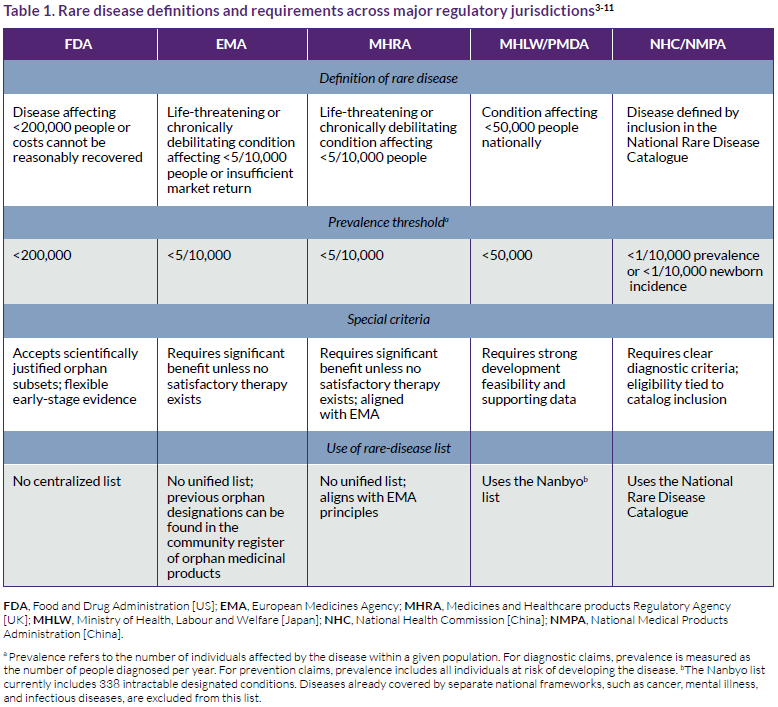
As a result of these differing definitions and criteria, the same condition may qualify as rare in one jurisdiction but not in another, independent of epidemiology alone. These differences establish the regulatory context within which orphan designation decisions are made.
Regulatory approaches to defining rare diseases
Across jurisdictions, regulatory authorities employ differing conceptual approaches to defining rare diseases. These include acceptance of orphan subsets, requirements for comparative benefit, feasibility expectations, and reliance on formal disease catalogs (Table 1). While prevalence thresholds determine formal eligibility, these embedded regulatory concepts play a more decisive role in designation feasibility.
Implications of divergent regulatory definitions
In the US, the Food and Drug Administration (FDA) emphasizes developmental flexibility by permitting orphan subsets and accepting limited early‑stage evidence, enabling earlier designation and regulatory engagement.3,8 In contrast, the EU and the UK introduce comparative evidence requirements at the designation stage through the significant‑benefit criterion, increasing evidentiary expectations earlier in development.4,5 In this context, the significant‑benefit criterion requires that, where a satisfactory method of diagnosis, prevention, or treatment has already been authorized, the sponsor demonstrates that the designated product is expected to confer a clinically relevant advantage or make a major contribution to patient care.4,5
Japan’s framework requires a theoretical rationale for the use of the product for the target population, and an appropriate development plan.6,7 This should be supported by existing nonclinical and Phase 1/2 data, unless the product is approved overseas or sufficient clinical study data are available. In China, the latest definition of rare disease allows for the inclusion or removal of conditions in China’s National Rare Disease Catalog. However, this list can constrain early regulatory engagement for newly characterized or ultra-rare conditions that have not yet been formally recognized.9-11
These regional structural differences influence not only whether a condition qualifies for orphan designation, but also the timing of applications, the nature of evidence generated, and the sequencing of regulatory submissions across regions.
Incentives, market exclusivity, and pathways
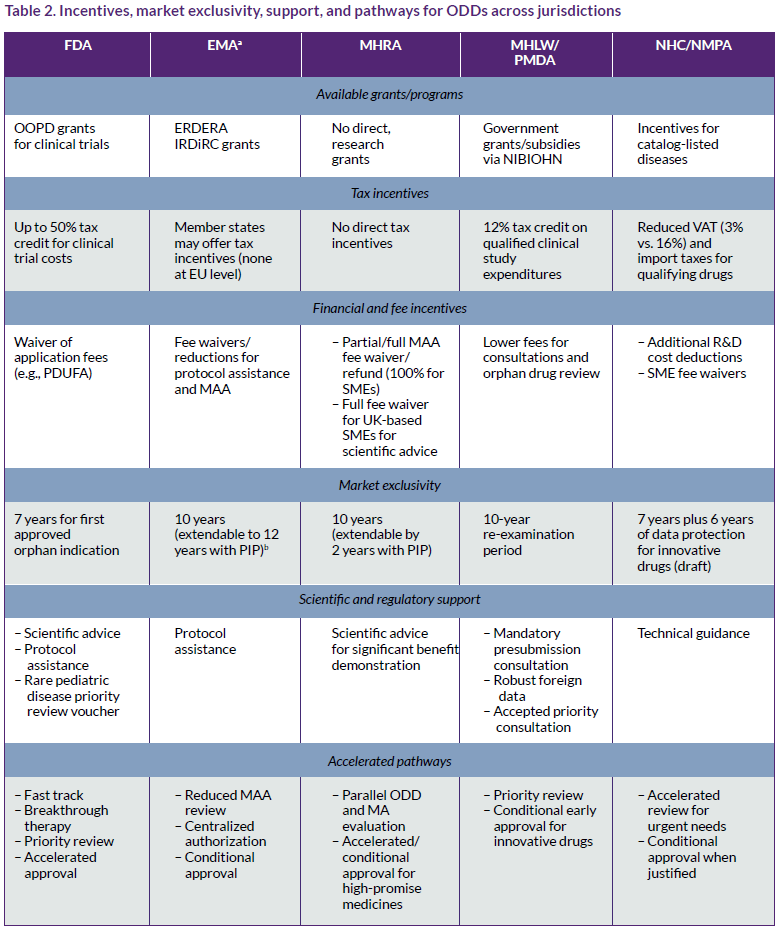
Beyond the differing definitions and requirements of ODDs shown in Table 1, ODD frameworks across jurisdictions also vary widely regarding the incentives offered, market exclusivity periods, regulatory support offered, pathways, and official registers (Table 2). It should be noted that some EU incentives and exclusivity provisions may change under the new EU General Pharmaceutical Legislation.

Proposed changes in the EU
The proposed EU General Pharmaceutical Legislation, which includes a new directive on the EU code and a new regulation laying down EU procedures, would introduce several changes affecting orphan designation, regulatory decision making, and market exclusivity.12,13 Under the proposed legislation, the orphan designation criterion based on return on investment would be removed; however, the practical effect is expected to be limited, as EU orphan designation has relied mainly on the prevalence threshold and the no satisfactory method/significant-benefit criterion.
The draft legislation would shift final decision making on orphan designation from the European Commission to the European Medicines Agency (EMA). Under the legislation, decisions on granting, refusing, or transferring orphan designation would no longer follow the current model, in which the Committee for Orphan Medicinal Products (COMP) adopts an opinion, and the European Commission issues the final decision. This change is linked to the broader reform of EMA committee structures, including the replacement of the current committee’s configuration by new working groups.
The proposed reform would also change how orphan market exclusivity operates in the EU:
- A company would no longer receive separate orphan exclusivity periods for multiple marketing authorizations covering the same active substance;
- An additional 12 months of exclusivity could still be earned if the product later obtains approval for a new orphan indication in a different orphan condition, although this extension would be limited and granted only under specified conditions; and
- Applications for similar products, including generics and biosimilars, could be submitted and reviewed before the orphan exclusivity period has fully expired, when less than two years of exclusivity remain, which would reduce the practical period of protection.
Another proposed change concerns the validity of orphan designation, which currently has unlimited validity, but under the new rules would expire after seven years. For existing orphan products, this validity period would begin when the regulation comes into force. Based on the current implementation timetable, the adopted acts are expected to enter into force in December 2026, with a transition period running to 2028; accordingly, for existing orphan products, this change would be expected to start applying from 2028. The validity may be extended upon a justified request if the sponsor can provide evidence that the relevant studies supporting the use of the designated orphan medicinal product in the intended conditions are ongoing and promising for filing a future marketing authorization application.
This proposed change is intended to encourage faster development and authorization of designated orphan products; however, it could also lead companies to apply for orphan designation later in the development process. In practice, a later application may allow sponsors to preserve more of the designation validity period closer to the time of marketing authorization, but it may also reduce the opportunity to benefit from orphan-specific incentives, such as protocol assistance, during earlier stages of development. To increase predictability for developers, the possibility of reviewing the eligibility criteria for market exclusivity after six years of the marketing authorization will be abolished once the proposed regulation is in force.12
Overall, these changes reduce the relative attractiveness of orphan designation in the EU compared with previous frameworks and other major jurisdictions because they narrow the practical benefit of exclusivity, limit lifecycle advantages across multiple orphan indications, and may weaken incentives for early designation.
China and Japan
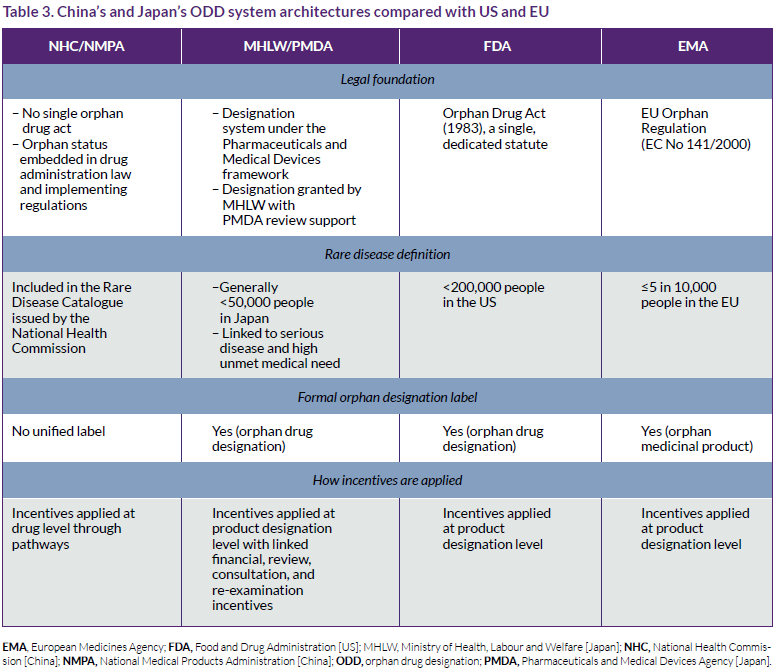
China’s system is functionally similar to those of the US and the EU in terms of outcomes but differs in structure (Table 3). China has no single Orphan Drug Act; instead, it operates a rare-disease drug framework built upon the National Rare Disease Catalog, accelerated regulatory pathways, and market exclusivity. In effect, China recognizes orphan drugs, but eligibility is not tied to a named ODD label.
China’s Drug Administration Regulations were revised in 2026.1 One of the new elements relates to eligible treatments of rare diseases, allowing for the possibility of a market exclusivity period of up to seven years. This possibility is contingent upon the marketing authorization holder (MAH) committing to a guaranteed drug supply. If the MAH fails to fulfill its supply obligations, the exclusivity will be revoked.14
In contrast, Japan applies a formal ODD, which is granted under the MHLW/PMDA framework, generally for serious conditions affecting fewer than 50,000 Japanese patients and supported by evidence of development feasibility, clinical need, and population relevance.6,7 Japan also offers a structured package of incentives linked to designation, including subsidies, tax credits, priority consultation and review, and a re-examination period that functions as postauthorization protection.6,7
Table 3 compares China and Japan’s ODD structures with those of the US and the EU. A UK comparison was intentionally omitted due to its similarity with the EU.
Transparency
To determine where to apply for orphan designation and the sequence of designations, manufacturers should consider transparency regarding the submitted data as a key factor.
A key difference between the EU and the other regions is that an orphan designation request application with minimal redactions can be made available to interested parties. The EMA’s policy on access to documents describes the rules the agency applies when granting access to documents it holds on human and veterinary medicines and documents not related to medicines.15 This can include documents produced by the EMA (e.g., opinion on orphan designation, COMP summary report, written comments from COMP members, and lists of questions) as well as the scientific part of the application prepared by the applicant.
Although the EMA allows limited redaction of commercially confidential information, this is a concern for some companies.16 Public disclosure may expose elements of a sponsor’s prevalence calculations, including how data sources are combined and whether commercial registries are used, as well as the planned approach for generating comparative evidence to support significant benefit at the time of marketing authorization. While the EMA permits redaction, sponsors may need to justify each proposed redaction under the applicable legal exceptions. This can be resource-intensive, and in some cases, proposed redactions are not accepted.
In the US, it is possible to request information on orphan designations from the FDA under the Freedom of Information Act, and indeed, in some cases, orphan drug designation requests have also been made.17 However, contrary to the EMA, large portions of these orphan designation requests are often heavily redacted by the FDA due to confidentiality protections. In Japan, China, and the UK, the data supporting orphan designation applications are not made public or are disclosed only after the marketing authorization is granted.
Cell and gene therapies
Cell, gene, and combined ex vivo gene-modified cell therapies (CGT) are eligible for orphan incentives across the major jurisdictions, but they are not framed identically in regulatory law.4,6,7,18-20 In Japan, cell and gene therapies may fall within the dedicated category of regenerative medical products under the Pharmaceuticals and Medical Devices Act, to which orphan designation can apply.6,7,19 In contrast, the US and China generally regulate these products within drug/biologic pathways, while the EU classifies many of them as advanced therapy medicinal products (Table 4).4,18,20
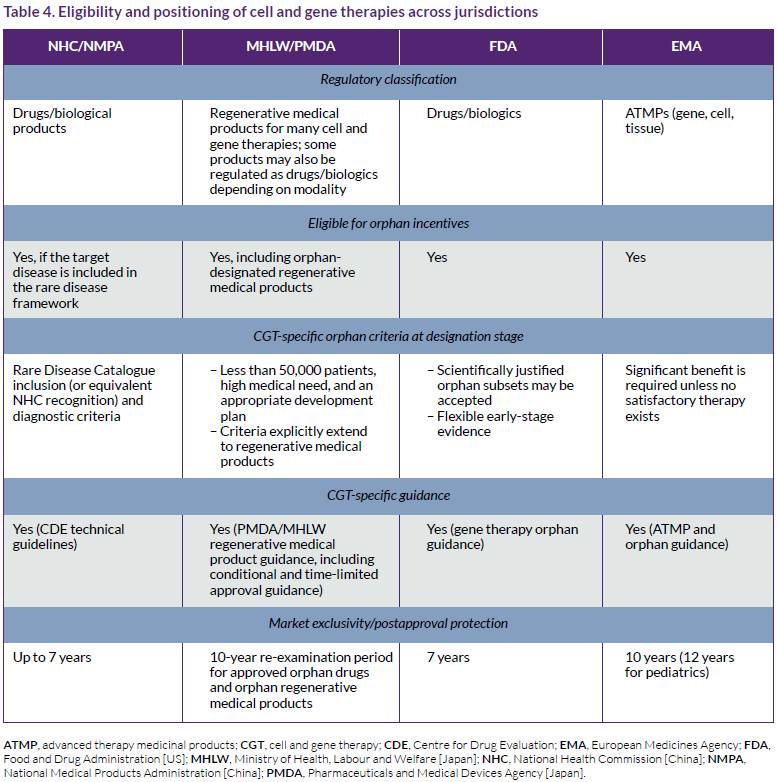
As shown in Table 4, the way cell and gene therapies are classified and incorporated into orphan-drug frameworks is broadly similar across regions. However, Japan has a distinct regenerative medicine framework, and China remains more dependent on rare-disease catalog recognition for orphan-linked positioning.6,7,9-11,19,20 The table does not include the UK, which has a very similar system to the EU.4,5,21
A practical global roadmap
A practical global orphan-drug roadmap should align the timing of designation requests, the design of evidence packages, and regulatory engagement plans with the distinct requirements of each jurisdiction.
- Sequencing: Begin in flexible jurisdictions (i.e., US, Japan) to secure early designation, then prepare stronger comparative evidence for the EU/UK. In addition, EU transparency and the possibility that orphan designation would remain valid for seven years (unless extended) support applying later in the EU.
- Evidence modularity: Build modular packages (epidemiology, diagnosis validity, natural history, comparative benefit, and bridging data) to meet different regional requirements.
- Policy resilience: Plan for evolving exclusivity rules and regulatory reforms (notably in the EU and China) by incorporating scenario planning early.
In practice, this means using early regulatory interactions in the US and Japan to establish feasibility and development momentum, while preparing the stronger comparative and diagnostic evidence needed for the EU, the UK, and China. A roadmap built on sequencing, evidence modularity, and policy resilience is more likely to preserve optionality across regions and reduce the risk that a product qualifies for orphan incentives in one jurisdiction but not in another.
Conclusion
No globally harmonized definition of rare disease exists, and orphan drug designation outcomes are primarily shaped by jurisdiction‑specific regulatory constructs rather than prevalence thresholds alone. The most influential divergence occurs at the designation stage, where Europe’s significant‑benefit requirement and China’s catalog‑anchored eligibility introduce early comparative and diagnostic constraints not observed in the US or Japan.
Over the past five years, orphan drug development has remained a major driver of regulatory innovation in the US, while China has demonstrated rapid alignment through regulatory reform, catalog expansion, and growing acceptance of multiregional clinical data. Japan continues to offer substantial lifecycle value for orphan products when feasibility and population-specific planning are addressed early.
As regulatory harmonization remains unlikely in the near term, effective global orphan strategies combine region-specific planning, including flexible early-stage development, comparative evidence generation, and alignment with local regulatory requirements. Differences in orphan drug designation frameworks also support a region‑specific sequencing strategy, particularly regarding the timing of designation applications and evidence development.
About the authors
Adriaan Fruijtier, MSc, is director of regulatory affairs at CATS Consultants GmbH with more than 37 years of regulatory affairs experience. His specialties include EU orphan medicinal product designation requests, pediatric investigation plans, scientific advice/protocol assistance requests, and MAA submissions in the EU. Fruijtier holds a degree in pharmacy from the University of Utrecht. He can be reached at [email protected]
Mauro Placchi, MSc, is a clinical development consultant with more than 30 years of regulatory and clinical development experience. His specialties include orphan drug development, regulatory strategy, protocol development, study management, and publication planning across pharmaceuticals, biologics, and medical devices. Placchi holds a degree in industrial chemistry from Milan University, Italy; a Certificat d’Etudes Complémentaires en Biophysique from Liege University, Belgium; and completed a fellowship at Brussels Free University, Belgium. He can be reached at [email protected]
Acknowledgment The authors thank Richard Phillips, MBBS, DPM, MBA (Canterbury, UK), for invaluable advice, and acknowledge the regulatory agencies and research organizations whose publicly available guidance documents, policy statements, and databases made this comparative analysis possible.
Citation Placchi M, Fruijtier A. Global regulations governing orphan drug designation. RAPS Journal of Regulatory Affairs. 2026;1(4):4-12. Published online 1 July 2026. https://www.raps.org/resource/global-regulations-governing-orphan-drug-designation.html
References
All references were last checked and verified on 12 June 2026.
- Wang CM, et al. Operational description of rare diseases: A reference to improve the recognition and visibility of rare diseases. Published December 2024. Accessed 5 June 2026. https://doi.org/10.1186/s13023-024-03322-7
- Haendel MA, et al. How many rare diseases are there? Nat Rev Drug Discov. Published 5 November 2019. Accessed 22 April 2026. https://doi.org/10.1038/d41573-019-00180-y
- Food and Drug Administration. Designating an orphan product: Drugs and biological products. Content current as of 12 August 2024. Accessed 22 April 2026. https://www.fda.gov/industry/medical-products-rare-diseases-and-conditions/designating-orphan-product-drugs-and-biological-products
- European Medicines Agency. Orphan designation: Overview. Not dated. Accessed 22 April 2026. https://www.ema.europa.eu/en/human-regulatory-overview/orphan-designation-overview
- Medicines and Healthcare Products Regulatory Agency. Rare therapies and UK regulatory considerations. Published 2 November 2025. Accessed 22 April 2026. https://www.gov.uk/government/publications/rare-therapies-and-uk-regulatory-considerations/rare-therapies-and-uk-regulatory-considerations
- [In Japanese] Ministry of Health, Labour and Welfare. Overview of the designation system for orphan drugs, orphan medical devices, and orphan regenerative medicine products. Not dated. Accessed 14 May 2026. https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000068484.html
- Sakushima K, et al. Orphan drug designation and development in Japan: 25 years of experience and assessment. Nat Rev Drug Discov. Published 15 March 2021. Accessed 22 April 2026. https://doi.org/10.1038/d41573-021-00045-3
- Food and Drug Administration. Frequently asked questions (FAQ) about designating an orphan product. Content current as of 11 May 2023. Accessed 22 April 2026. https://www.fda.gov/industry/designating-orphan-product-drugs-and-biological-products/frequently-asked-questions-faq-about-designating-orphan-product
- Lu Y, Han J. The definition of rare disease in China and its prospects. Intractable Rare Dis Res. Published 28 February 2022. Accessed 22 April 2026. https://doi.org/10.5582/irdr.2022.01034
- National Health Commission of the People’s Republic of China. China issues new rare disease guidelines [press release]. Published 11 July 2025. Accessed 22 April 2026. https://en.nhc.gov.cn/2025-07/11/c_86493.htm
- The State Council of the People’s Republic of China. China releases second catalog of rare diseases [news release]. Updated 21 September 2023. Accessed 22 April 2026. https://english.www.gov.cn/news/202309/21/content_WS650b7dc6c6d0868f4e8df9f1.html
- Council of the European Union. Regulation laying down Union procedures for the authorisation and supervision of medicinal products for human use and establishing rules governing the European Medicines Agency [analysis of the final compromise text]. Dated 24 February 2026. Accessed 22 April 2026. https://data.consilium.europa.eu/doc/document/ST-6366-2026-INIT/en/pdf
- Proposal for a Directive of the European Parliament and of the Council on the Union code relating to medicinal products for human use, and repealing Directive 2001/83/EC and Directive 2009/35/EC. Dated 26 April 2023. Accessed 31 May 2026. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:52023PC0192
- [In Chinese] Regulations for the Implementation of the Drug Administration Law of the People's Republic of China (Decree No. 828 of the State Council of the People's Republic of China). Published 28 January 2026. Accessed 2 May 2026. https://www.mee.gov.cn/zcwj/gwywj/202601/t20260128_1142757.shtml
- European Medicines Agency. European Medicines Agency policy on access to documents. Published 4 October 2018. Accessed 22 April 2026. https://www.ema.europa.eu/en/documents/other/policy-43-european-medicines-agency-policy-access-documents_en.pdf
- European Medicines Agency. Overview of comments received on European Medicines Agency policy on access to documents Policy 0043. Published 3 December 2018. Accessed 22 April 2026. https://www.ema.europa.eu/en/documents/comments/overview-comments-received-draft-policy0043-european-medicines-agency-policy-access-documents_en.pdf.
- Food and Drug Administration. FDA FOIA logs. Current as of 11 June 2026. Accessed 2 May 2026. https://www.fda.gov/regulatory-information/freedom-information/fda-foia-logs
- Food and Drug Administration. Cellular & gene therapy products. Current as of 11 January 2026. Accessed 13 May 2026. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products
- Pharmaceuticals and Medical Devices Agency. Regenerative medical products. Not dated. Accessed 13 May 2026. https://www.pmda.go.jp/english/review-services/reviews/0003.html
- National Medical Products Administration. Ciltacabtagene Autoleucel injection approved for marketing by China NMPA [press release]. Updated 19 February 2025. Accessed 13 May 2026. https://english.nmpa.gov.cn/2025-02/19/c_1073591.htm
- Medicines and Healthcare Products Regulatory Agency. Advanced therapy medicinal products: Regulation and licensing in UK. Published 26 January 2015. Accessed 13 May 2026. https://www.gov.uk/guidance/advanced-therapy-medicinal-products-regulation-and-licensing


