

Life after the FDA: Career paths for former regulators
While FDA experience is highly valued in the private industry, transitioning from the agency to private-sector employment requires new skills and a shift in mindset. This article provides resources for regulatory professionals with FDA experience seeking careers in the private sector. The article explores common career pathways in the medical device space, addresses key differences between the FDA and industry roles, and provides strategies for leveraging FDA experience outside the agency. The insights are based on the authors’ experiences after departing from the FDA. The article is divided into two sections – the first outlining career pathways and the second offering advice on job hunting.
Keywords – career options, FDA experience, industry
Introduction
Professional experience at the US Food and Drug Administration (FDA) is highly valued across the healthcare sector. While many former FDA employees find opportunities in traditional regulatory affairs (RA) or regulatory consulting roles, the possibilities are expansive. Transitioning to a position outside the FDA typically requires broadening one’s skillset. Jobs at the FDA primarily support regulatory activities related to medical products marketed in the US, whereas industry roles can require a broad understanding of the global regulatory landscape, including both pre- and postmarket considerations as well as quality management.
Although the FDA and the medical product industry share a common focus on public health and patient care, their roles differ. In particular, the world outside the agency is far from straightforward. Industry professionals must navigate a dynamic landscape where scientific, financial, and regulatory priorities intersect. The timing of market access has significant financial implications, making identifying the least burdensome regulatory pathway a critical priority. The submissions the FDA receives are generally the final, polished outcome of an intricate process involving significant internal deliberation, decision making, and refinement within companies. Meanwhile, the full medical product development lifecycle, most of which does not directly involve the FDA, is iterative, nonlinear, and fraught with uncertainties. Effective risk assessment and management are central to product and project development, adding another layer of complexity to industry roles.
Creativity, flexibility, and the ability to leverage existing knowledge and skills are valuable qualities in a competitive market. Understanding the variety of private-sector job opportunities can help professionals with FDA experience identify where their experience can best transfer and have an impact. In addition to knowledge of the US medical product regulatory framework and familiarity with cutting-edge technology, FDA experience typically gives employees excellent technical, project management, communication, and negotiation skills that they can likely leverage for industry roles.
Staffing the FDA with top experts is paramount to the entire healthcare system, including patients, providers, and the medical product industry, who all benefit from efficient and scientifically sound review processes. While the agency offers unique career experiences and benefits specific to public service, an individual employee’s preferences, needs, geographic constraints, or external circumstances, such as shifts in agency priorities, can motivate or require them to consider careers outside the agency.
The article is based on the collective knowledge and experience of former FDA personnel who moved into various professions within the healthcare sector before the recent large-scale departures from the agency. It presents common post-FDA career pathways, discusses the differences between the FDA and the private sector, and offers advice on identifying and applying for opportunities outside the FDA. Although the article focuses mainly on the authors’ expertise in the medical device industry, most of the information in it can also apply to the pharmaceutical and biologics sectors.
Section 1 – Career pathways
Former FDA employees can assume a range of roles after their time at the agency. Many find employment in regulatory affairs or quality assurance, either in-house at a company or in consulting. Regulatory knowledge is necessary throughout the product development cycle, so former FDA employees may also branch into various other roles related to clinical trials, medical writing, new product development, software and cybersecurity, project management, or marketing. This section discusses the general scope of duties common across regulatory roles and outlines career paths to which FDA skills may transfer.
Medical device manufacturers or distributors
Many FDA staff find employment at medical product manufacturers or distributors. These companies range from early-stage start-ups with few employees to multinational corporations (MNCs) several times larger than the FDA to nontraditional medical product manufacturers such as big tech companies. Most of the companies in the medical device industry are relatively small. One study that analyzed economic data from the Census Bureau found that 73% of medical device firms had fewer than 20 employees.1
Although this section focuses on traditional regulatory affairs roles, former FDA employees may also find careers in various other industry functions, including medical affairs, clinical affairs, quality assurance, and research and development (R&D) – a common pathway for former FDA research scientists.2
Scope of duties
The general scope of a regulatory professional’s duties at medical device manufacturers and distributors is roughly similar across companies and organizations, regardless of size, and falls into the following general categories with their respective responsibilities:
Regulatory strategy and compliance
- Develop and execute regulatory strategies to bring products to the commercial market. FDA experience, which involves exposure to a large volume of regulatory submissions, is valuable in many aspects of strategy development, including identifying potential labeling claims and determining their alignment with planned verification and validation.
- Ensure compliance with applicable statutes, regulations, and standards. In addition to being familiar with the Quality System Regulation (QSR; 21 CFR 820),3 the EU Medical Device Regulation (EU MDR),4 and EU In Vitro Diagnostic Regulation (EU IVDR),5 regulatory professionals working with devices should be familiar with ISO 13485,6 ISO 14971,7 ISO 62304,8 and other relevant standards. These standards may not always be relevant to FDA reviews but are of great practical importance in industry. It is particularly important to understand design controls.
- Ensure sponsor readiness for bioresearch monitoring for the smooth operation of clinical trials and good laboratory practices, compliant with applicable regulations (21 CFR Part 50 – Protection of Human Subjects,9 21 CFR Part 54 – Financial Disclosure,10 21 CFR Part 56 – IRBs,11 21 CFR Part 58 – Good Laboratory Practice for Nonclinical Laboratory Studies,12 21 CFR Part 809 – In Vitro Diagnostic Products for Human Use,13 and 21 CFR Part 812 – Investigational Device Exemptions).14
- Understand the regulatory risk profiles of target companies through regulatory due diligence when considering investments, acquisitions, mergers, or partnerships.
Regulatory submissions
- Lead preparation and submission of regulatory filings. For medical devices, this includes 510(k)s, de novos, premarket approval applications (PMAs), investigational device exemptions (IDEs), Q-submissions, breakthrough device interactions, and technical documentation for CE marking. As submissions require significant cross-functional resources to prepare, unlike FDA reviewers, an industry regulatory professional sees fewer submissions and has duties other than filing regulatory submissions.
- Respond to regulatory agency inquiries and deficiency letters.
- Maintain submission timelines, track regulatory approvals, and communicate with internal stakeholders because marketing and sales activity must be planned around marketing authorization.
- Assess product changes for regulatory impact and determine any changes in submission requirements. The FDA does not generally see letters to file (LTF) unless they are requested during an inspection or premarket review. Meanwhile, in industry, regulatory professionals are often responsible for determining whether changes are appropriate for an LTF or whether they require a regulatory submission. Depending on the technology and quality management system, drafting LTFs can be as lengthy and involved as actual regulatory submissions. Just as obtaining FDA marketing authorization is a required step before certain products can be commercially released, for changes to products made by LTFs, the LTF must be finalized and approved by all internal stakeholders before a modified product can be marketed. It is important to complete LTFs promptly and ensure that the changes they describe have been adequately assessed.
Quality system and risk management
- Collaborate with quality teams to ensure regulatory compliance with design controls, risk management (ISO 14971), labeling, unique device identifier (UDI), and postmarket surveillance.
- Support audits and inspections conducted by regulatory agencies and notified bodies.
- Develop and review regulatory policies and standard operating procedures (SOPs).
Cross-functional collaboration
- Provide regulatory guidance to R&D, clinical, and quality teams during product development; this might include guidance on indications, verification, and validation strategy.
- Work with marketing to review and approve labeling, advertising, and promotional materials that comply with regulatory requirements. At some companies, advertising and promotional activities, known as ad promo, can be a dedicated function.
Regulatory intelligence
- Proactively monitor the competitive market landscape.
- Monitor, analyze, and support compliance with new and modified global regulations and standards.
- Present changes in regulations and standards to cross-functional management teams with executive responsibility during the management review. Regulatory professionals also provide options for organizational investments and timelines to comply with changes.
Postmarket activities
- Oversee regulatory reporting requirements, including medical device reports, vigilance reports, and field safety corrective actions (FSCAs).
- Support product lifecycle management, including regulatory updates and renewals.
Regulatory roles at medical product distributors differ from those at manufacturers, even if the distributor and manufacturer both fall under the same corporation. For example, a US distributor subsidiary may handle FDA submissions and US postmarket compliance for an overseas subsidiary manufacturing devices within a single corporation. In this example, the US distributor subsidiary does not manufacture devices. It may not have a quality management system or an in-house R&D team, resulting in a more limited scope of regulatory duties.
Multinational corporations
MNCs may have dedicated roles for specific regulatory activities. These roles can be divided into pre- or postmarket, intelligence, advocacy, and advertising and promotion functions. MNCs are typically publicly traded, requiring them to report financial results quarterly. As a result, the end of each quarter is often a particularly important time.
MNCs may have multiple, entirely separate regulatory teams corresponding to different business units, subsidiary companies, or diagnostic/therapeutic/medical specialties within the corporation that combine similar product knowledge with regulatory strategy. The level of specialization varies across teams. In addition to portfolio-specific teams, MNCs also have enterprise-level regulatory units that provide shared resources to drive efficiency in common needs for regulatory strategy, policy development or shaping, and operations. Ultimately, organizational structure is informed by market complexity, geographic distribution, product portfolio, and corporate culture. There are two common approaches by which MNCs organize their core regulatory structures:
- Total product lifecycle (TPLC) approach – In this approach, also known as cradle-to-grave, a regulatory professional will be responsible for a product throughout its lifecycle. This includes being responsible for the new product development (NPD) process for opportunity/feasibility assessment, developing regulatory qualification strategies with cross-functional teams, and understanding jurisdictional requirements such as market-specific labeling and local testing. The TPLC approach also includes regulatory needs in the postmarket sustaining process, which is focused on maintaining product approvals in regulated geographies, assessing product iterations for regulatory impact, and expanding patient access through indication expansions. This approach requires a regulatory professional to have a solid understanding of design controls and cross-functional collaboration skills. Responsibility by region will vary by MNC, and MNCs often use local market representatives to support an activity when it is not the direct responsibility of the regulatory professional otherwise responsible for that product.
- Regional approach – In this approach, regulatory responsibilities are split between regional teams (Africa, Asia, Europe, Latin America/Caribbean, Middle East, North America, Oceania, Europe, and Asia) and a global team. Regional regulatory professionals are responsible for market access and maintenance activities, while global regulatory professionals are responsible for new product development and postmarket activities, such as recalls or field actions.
Due to the size and complexity of MNCs, regulatory professionals should have (or be able to gain) a solid understanding of the responsibilities of the cross-functional partners within an organizational structure where multiple teams frequently collaborate and share responsibilities. As these partnerships are not as intrinsically linked as they would be in smaller organizations, regulatory professionals need effective collaboration and consensus-building skills.
Beyond the identified core structures, MNCs often have regulatory functions that ensure the company keeps up to date on new and changing regulatory information. These functions may be identified as regulatory policy and intelligence. An MNC may have one or both functions depending on its organizational structure. The regulatory policy function partners with external trade associations and health authorities to ensure the company remains aligned with current thinking and to advocate for positions that best serve the interests of the company and its patients. This function also partners internally to identify strategic priorities, align on external communication needs, and keep relevant partners up to date on advocacy activities and learnings. The regulatory intelligence function proactively monitors the competitive landscape while monitoring, analyzing, and supporting compliance with new or changing regulations and requirements. These activities are critical to ensure a company is in a continual state of compliance with an ever-changing regulatory environment and that new products are developed and brought onto the market with all requirements considered.
The work that regulatory professionals perform at MNCs complements FDA efforts. This is particularly true of the premarket submission focus, where regulatory affairs professionals work closely with cross-functional partners to compile the necessary components of a submission and weave them into a concise, logical case for the product’s safety and effectiveness that the FDA will ultimately review.
Midsized companies
A midsized company is typically defined as falling between a small business and an MNC in terms of revenue, number of employees, and market reach. Definitions vary, but midsized companies generally have annual revenues between 50 million and 1 billion and between 100 and 1,000 employees.15,16
RA professionals at midsized companies often have a broader scope of responsibilities than their counterparts in MNCs. Due to leaner teams, individuals may handle multiple regulatory functions and global regions. At MNCs, regulatory roles are typically more specialized, and distinct teams may manage different functions.
Start-ups
Start-ups are generally cutting-edge, developing first-of-their-kind products. In the US, novel medical devices may be subject to de novo classification or PMA. Start-ups may offer experience in preparing Q-submissions, designation requests for breakthrough devices, IDEs, and other submissions appropriate for novel, early-stage products. Because test methods for novel devices are not always well established, start-ups may offer more iterative interactions with the FDA than companies with more established technologies.
Compared with regulatory professionals at larger organizations, those at start-ups often take on multiple roles, sometimes combining regulatory responsibilities with quality, clinical, or other functions. With fewer hierarchies between employees and executive leadership, decision making tends to be more agile, and ideas and strategic pivots are implemented more rapidly. However, informational infrastructure, such as SOPs and document templates, may need to be established from scratch. In this sense, a start-up is the opposite of the FDA, where procedures for regulatory review are generally well established, and efforts focus on the review itself.
Relative to larger companies where senior regulatory staff may have decades of experience within the company or similar companies, regulatory expertise within a start-up may be limited. In the early years of a start-up, the regulatory burden associated with bringing a product to market may be significantly underestimated. Hires in engineering or project management may come from industries outside of life sciences. These hires are sometimes unfamiliar with the specifics of design controls, benefit-risk-based decision making, and FDA interaction. Regulatory professionals at start-ups have fewer colleagues to serve as back-ups and must often rely on external resources for guidance or mentorship. Some start-ups may lack any experience with regulatory submissions altogether. In many cases, regulatory functions are initially outsourced to consultants, with full-time regulatory professionals brought on board only when the company reaches a more advanced stage of development.
Start-ups are typically funded by angel investors, venture capital, or grant funding. Compared with larger corporations, start-ups generally offer less stability, as the viability of the company is dependent on successfully raising each round of funding. Compensation packages, including benefits and leave policies, may be less competitive than at larger companies. The working space and facilities may be bare-bones, and workloads can be unpredictable or heavier than in established companies. Stress levels can be significant, particularly in single-product start-ups whose company's viability depends on securing regulatory marketing authorization before funding runs out. Job titles may not align with traditional corporate structures—even senior positions, such as vice president or global head, may come without direct reports. In exchange for taking on a unique set of challenges and operating in a resource-constrained, high-intensity environment, start-ups often provide equity grants as part of their compensation package. When considering compensation packages, equity is comparable to a lottery ticket; while the equity could have tremendous value someday, the more likely outcome is that it will have little or none.
In many respects, start-ups are very different from the FDA. For former FDA employees who can tolerate a degree of uncertainty and ambiguity and find high-risk/high-reward scenarios appealing, start-ups present unique opportunities for career acceleration and skill development. FDA experience in project management, communication and negotiation, cross-functional leadership, technical writing, and regulatory science research, as in other types of companies, can directly translate to various roles at a start-up. Due to the critical nature of their work, regulatory teams often have high visibility within the company. Given their focus on cutting-edge technologies, start-ups can be particularly well-suited for PhD-trained regulatory professionals, as their research experience can help them engage closely with R&D and clinical teams and play an integral role in shaping validation strategy and cross-functional decision making.
Consulting
Regulatory consultants can take on a range of responsibilities across the industries they serve – from navigating the complex regulatory landscape to identifying compliance gaps with regulation to preparing documentation to submit to regulatory agencies. As is the case for many regulatory professionals, consultants typically encounter a wide range of products. Therefore, it may be less important for consultants to have specific product expertise than having the ability to take initiative and a willingness to learn. On the other hand, familiarity in areas such as artificial intelligence and machine learning (AI/ML), biocompatibility, sterility, cybersecurity, and statistics are applicable to many products. Familiarity with FDA constructs, such as certain statutes, regulations, and regulatory processes, would serve a professional well because the concepts span all product types.Although consultancies vary in size, one thing is constant – billable hour requirements. Billable hours are how firms bring in revenue. If one were to bill seven and a half hours a day for five days a week, that would result in 37.5 hours a week. Considering that, on average, there are four weeks in a month, the month’s total billable hours would be 150, or 1,800 hours per year, assuming that no vacation, holidays, or sick days have been taken. (For reference, the law firms’ average billable hours per year range from 1,700 to 2,300). This also does not consider time taken for lunch, short breaks, reviewing the trade press and general correspondence, or attending internal meetings or conferences. Keep in mind that commuting time, interviewing potential candidates, training, and other activities have also not been factored in. Therefore, a consultant with seven and a half billable hours per day could easily spend at least 10 hours working.
Across firms, compensation is generally composed of salary, bonuses, and profit sharing. When considering a consulting position, it is critical to understand whether the stated billable hour numbers are an average, target, or minimum and if there is any impact on performance reviews and bonuses if the hours are exceeded or not met. A utilization rate is often a tracked metric in consulting groups and management consulting firms and is generally calculated as follows:
(Billable hours / total hours) x 100 = utilization rate
For example, if a professional bills 36 hours and is expected to work 40 hours a week, their utilization rate is (36 / 40) x 100 = 90%. A high utilization rate indicates that a professional maximizes their time on revenue-generating activities. It is also a metric to determine profitability and reflect resource optimization.
While these overarching principles are nuanced, it is important to note that billable work will be the key consideration in success as a consultant, whether the role is part of a consulting group, in an independent capacity, at a law firm, or within a management consulting firm.
Independent contractor/consultant
Providing guidance and expertise as an independent contractor can be one of the most flexible arrangements in regulatory affairs with regard to the types of projects, clients, and strategies used to address a regulatory need. Independent contractors can advise over a range of general regulatory disciplines and across many product types or focus on a specific technical area of expertise, for example, supporting software or cybersecurity efforts, standalone and clinical performance assessment, biocompatibility, or sterilization. Clients can range from start-ups to MNCs with unique needs suited for different types of consultants. Support can include developing a regulatory strategy for a new product, outlining an effective communication strategy with a regulatory authority, remediating documentation intended for a premarket submission, or providing regulatory assessments as part of due diligence efforts for an investment group, among many others. Successful independent contractors can stretch their technical and business acumen to meet client needs and deliver sound advice, guidance, and work products to help them bring safe, effective, and innovative products to market.
To identify their place within the regulatory space, independent contractors must consider where and how they would like to contribute their expertise. Many independent contractors work solely within their own company on specific projects, which are managed and facilitated internally. However, contractors can also choose to work on projects managed by other independent contractors or consulting firms. Depending on the type of work, many contractors combine these arrangements to focus on their desired projects and networks.
Unlike employment within an established company, independent contractors have considerable administrative responsibilities, such as managing legal and regulatory compliance related to the business, including registrations, reporting, taxes, and payments. The legal and tax implications and requirements will depend on the structure of the business, most of which will operate as a limited liability company (LLC) reporting as a sole proprietor or S-Corporation. Articles of incorporation and annual reporting requirements will vary based on this structure but are always filed with the state where the business is incorporated. In addition, IT system set-up and management, including email, website, and data back-up and security, are necessary tasks.
Like any small business owner, independent contractors must navigate the accounting and invoicing processes to achieve a successful career. There are accounting, time tracking, and invoicing systems, often integrated with each other, that can streamline the tracking of generated income, expenses, and other payments or contributions related to one’s business. A tax professional can also work with these systems to ensure there is accurate reporting for tax filings each year. Independent contractors can choose how to track and invoice the costs associated with their services, varying from project-based fees to fees generated from billable hours accrued while working on a specific project or activity.
Professional liability insurance is strongly recommended to protect the business if the contractor is sued for negligence in delivering consulting services. This insurance typically covers the amount from one occurrence or a total aggregate amount to defend against a lawsuit judgment or settlement. Other insurance coverage, such as insurance related to cyber risk, liability, and data breaches, is also encouraged in the current cyber risk landscape. Regardless of the insurance coverages and policies in effect, it is important to mitigate as many risks as possible at the outset through robust contracts and secure systems.
To this end, independent contractors must also be familiar with contracts and legal language as clients are acquired, projects are outlined, payment terms are chosen, and deliverables are mutually defined to ensure that expectations are clear. Obtaining legal counsel with expertise in business law is advised to help draft contract templates and review incoming contracts and agreements from potential clients and firms. Establishing a close working relationship with counsel can facilitate timely review of these documents, leading to efficient onboarding and project kick-off. Large companies have extended processes to onboard new external contractors, including compliance efforts on anti-bribery, anti-kickback, and other anti-corruption policies that are required for corporate governance. Therefore, working for an already-approved vendor may shorten the onboarding process and allow the contractor to begin work sooner.
Independent contractors must also be flexible about their finances because income may fluctuate each month as the number of projects changes, given availability or delays. These fluctuations can be mitigated by arranging for consistent income through retainer payments from clients (typically larger corporations) looking for interim but dedicated support over a set period. Additional financial stability can be achieved by collecting deposits before starting a project and setting rates that follow industry trends and align with the contractor’s market value. Despite these challenges, as the contractor’s client base grows, in addition to their understanding of industry standards of various service areas, their financial projections tend to become more predictable.
Finally, establishing a professional network is the most important step for independent contractors. Networking with regulatory professionals with similar or complementary expertise can lead to new relationships and partnerships. These partnerships can be with other independent contractors, consulting or law firms, academic institutions, and private sector companies. Setting aside time for business development and networking can create a healthy and continuous pipeline of projects.
Consulting groups
Companies of all sizes and stages of development often engage with consulting groups through referrals from their investor community, brand recognition, or previous interactions. Smaller boutique consulting firms focus on a singular area of expertise. They may choose to provide limited services, whereas larger consulting groups can sometimes serve as a one-stop shop for regulatory, compliance, clinical, and testing needs. Many consultancies offer flexible remote working arrangements and encourage travel for direct interaction with clients and business development opportunities.
The roles and responsibilities within a consulting group vary depending on the services offered. In larger organizations, consultants at all levels often focus on one aspect of the total product life cycle (e.g., premarket regulatory support, verification and validation (V&V) activities, or postmarket compliance activities) and work with one type of product (e.g., device, drug, biologic). As previously discussed, specialized consultants are sometimes brought in to provide professional services related to a specific technical area, such as AI/ML, cybersecurity, biocompatibility, or clinical support.
Regardless of the size of the consultancy, nearly all employees are required to engage in some form of business development by bringing in new clients, projects, and, ultimately, revenue. While some groups offer a set salary with no additional compensation for revenue generation, others may offer a lower salary with significant bonuses for bringing in new work or supplement managers’ salaries with a bonus based on meeting revenue targets. Unlike the flexibility offered as an independent contractor, consulting groups most often track billable hours as a measure of productivity. While the billable hour targets are different for each consultancy, as a rule of thumb, most aim for the revenue generated by a consultant’s billable work to be at least three times their salary to account for company costs and employee benefits. This can lead to target utilization rates greater than 80% to ensure that billable client work is sufficient to generate revenue that can cover the costs noted above.
Many consultancies use independent consultants who are not paid a set salary but are used instead as projects commence to extend their client support during temporary increases in workload. Standard independent consulting agreements define the relationship between the consultancy and the contractor and detail the terms of payment and engagement, along with intellectual property rights and confidentiality. This model works particularly well for large remediation projects, which require significant human capital to improve or overhaul corporate systems and processes but close after a set amount of time.
Law firms
Law firms offer regulatory professionals a unique opportunity to assist throughout the product lifecycle. Companies with FDA-regulated products may seek out law firms because they provide a one-stop shop for regulatory, compliance, transactional, and litigation issues. Professionals in this field may represent a client during a pre-submission meeting with the FDA, respond to Form FDA 483 observations, advise on mergers and acquisitions (M&As), and conduct a government investigation all in a single workday.
For those interested in working at a law firm, it is important to understand whether billable hours are specific to attorneys or non-attorney professionals. In addition, law firms typically expect their billable employees to engage in nonbillable activities, such as business development, training, and authoring publications. Given the globalization of companies, many professionals at law firms work with clients across all time zones, significantly extending their working hours. In addition, projects may have short turnaround times. Being flexible and understanding that work hours may not always be a traditional 9-to-5 schedule can help set realistic expectations for a work-life balance.
Law firms consist of partners, counsel, associates, non-attorney professionals, and support staff, all working together on different matters. Unlike other entities discussed in this article, law firms typically have fewer resources for regulatory and compliance matters. Where medical device start-ups may have around 20 employees focused solely on ensuring their product reaches the market, a law firm may have fewer than a handful of employees dedicated to helping one client. A professional’s experience and training on a given regulatory matter will depend on the culture of the law firm and practice group.
An attorney who worked at the FDA for several years may join a firm as counsel and be responsible for supervising other attorneys and non-attorneys. Given the relatively flat hierarchy, a non-attorney professional is unlikely to have supervisory responsibilities. However, both attorneys and non-attorneys at law firms demonstrate management skills, including time management, communications, strategy, and leadership, as they advise a client throughout the FDA submission process.
Working at a law firm can be very rewarding, given the heterogeneous nature of legal matters. Developing creative solutions for complex issues and working towards a positive outcome for various clients can be intellectually stimulating and professionally satisfying. Former FDA employees who thrive on intense engagement with diverse personalities and are open to working outside of regular hours may find that the challenges of working at law firms also come with unique benefits. Those who embrace these aspects will likely adapt quickly and thrive in this professional setting.
Management consulting firms
Law firms and management consulting firms (also known as professional services firms) share some similarities. For instance, both have specialties (e.g., health/life sciences) and capabilities (e.g., audits, M&As, risk management, regulatory compliance) within the regulatory space. Both may also focus on the federal government or state and local governments. Companies that have established relationships with management consulting firms for tax or accounting advice may find it convenient to extend those partnerships to include regulatory and compliance work.
Given the nature of management consulting work, firms may refer to their work as advisory. Advisory work encompasses a range of functions and capabilities that may someday shape industries and organizations. For instance, management consulting firms have supported IT modernization and changed the way the industry interacts with the FDA. They have also been tasked with improving business process and implementation support. Familiarity with the FDA systems and operations can help consultants inform and strategize with clients on the next best business action.
The expectation for utilization rates will vary depending on seniority at management consulting firms. As one progresses up the career ladder, there is less of a focus on utilization and more on business generation, which can include building client relationships, scoping out new opportunities, and drafting project proposals.
Another similarity between law firms and management consulting firms is the nonbillable expectations. For example, there may be an expectation to be in the office, complete training, and work with others on business development activities.
The organizational structure can vary slightly from firm to firm, but those with a few years of experience are generally considered associates or analysts. An associate or analyst can then progress to senior associate/consultant, manager, director, and partner. Professionals with an MBA or PhD may start as senior associates or consultants.
An associate or senior associate may have to analyze data and synthesize findings to issue recommendations. Those with a statistical, data science, or AI/ML background may find a role as a data scientist in a firm’s life sciences practice. Other projects focus on advertising and promotion, quality management systems, and cybersecurity.
Unlike at the FDA, where work is assigned to a reviewer, professionals at management consulting firms develop relationships with engagement partners and mentors to be staffed on projects. This ensures they are engaged in revenue-generating work, not “on the beach” (a consulting term for a temporary break in client projects). Projects can be months or even years long and require personnel to travel long distances to a site. As a result, professionals typically focus on a specific project during a given day.
The collaborative nature of advisory work is similar to the operation of teams within the FDA. For example, an FDA lead reviewer will work with consultants and issue a decision on a marketing submission, whereas an advisory professional will work with colleagues on a team. They conduct baseline assessments, perform analyses, and conduct research – all of which will be synthesized into results and findings for client management. During management presentations, the teams offer recommendations for action based on these findings. Therefore, prior experience as a reviewer working with others and a background in science, engineering, technology, and/or mathematics (STEM) is valuable, as work will likely be team-oriented and technical. Many job postings in the life sciences industry require a STEM background.
CROs and testing laboratories
Contract research organizations (CROs) and testing laboratories are important aspects of medical device development and commercialization. CROs are independent of medical device manufacturers and provide a wide variety of specialized services throughout the entire medical device life cycle. CRO services can be wide-ranging and include research and validation testing, preclinical animal testing, biocompatibility, microbiology and sterility, physicochemical testing, cybersecurity/software, imaging, and more. Many CROs are also involved in designing and managing clinical studies, including data management and statistics, quality assurance, regulatory and technical consulting, reimbursement aspects, medical writing, and many other areas. A larger CRO often has a variety of office spaces and testing laboratories to provide this broad range of services.As an independent entity, the CRO is often relied on for its experience and expertise with specific medical device types or technologies. As most medical device manufacturers do not have in-depth technical expertise, bandwidth, or facilities to perform medical device testing, working with a CRO or testing laboratory is usually necessary. CROs often have teams specializing therapeutic areas for medical devices and in vitro diagnostics and often see a large throughput of devices. CROs can help mediate discussions between the FDA and industry partners, as they typically have a favorable and unbiased relationship with both parties. Larger CROs may also have a global footprint, enabling manufacturers to develop plans and testing that simultaneously meet the needs of multiple markets.
Depending on their roles, employees of these larger CROs may work on site in the US, abroad, or remotely. Laboratory employees typically work on-site, while the work of most other employees can be performed remotely. Travel requirements also vary greatly depending on the employee’s role. For example, those working in laboratories may not have many travel requirements, while consultants may travel to client sites as requested or needed for specific projects. Management and sales positions and any that require on-site presence may be required to travel more extensively.
Depending on the project and employee role, projects may be fixed fee or hourly based. Like law and consulting firms, projects are subject to utilization rate requirements. The requirements may be less demanding than those of a law firm but will likely still be high. Utilization rate requirements often translate to less nontechnical work, such as internal development activities and travel to conferences.
Compared with independent consultants, CROs or consulting firms may offer less flexibility in choosing projects and clients may also have a heavier workload. At a CRO, consultants are often more limited in their range of work and will be required to focus on specific areas to meet higher utilization rates.
Working at a CRO can be a natural transition for former FDA employees who have worked with premarket medical device submissions or were engaged in clinical trials. The teams may be similarly structured in terms of subject matter expertise. The teams may also have similar roles, such as reviewing or drafting premarket submissions. Former FDA clinical or statistical experts may find similar roles at a CRO involving clinical trial design and management, conducting bioresearch monitoring audits, data analysis/management, and medical writing. These roles may include working directly with the client’s medical experts, institutional review boards (IRBs), and healthcare facilities. Technical experts may be relied upon to draft test strategies or justifications that the FDA will accept. They often work directly with the FDA’s technical experts to agree on a strategy for their client. As with working at the FDA, working at a CRO involves a range of technical expertise (e.g., veterinary medicine, clinical, biocompatibility, sterility, and statistics). Therefore, CRO professionals provide substantial support for their fellow employees and their clients.
International health authorities/notified bodies
FDA experience can provide professionals with many skills that can be leveraged at an International Health Authority (IHA). However, while benefit-risk assessment and quality systems experience may be directly transferable, addressing gaps in knowledge of regulatory frameworks and structures outside the US is critical. Some IHAs, like Health Canada and the Therapeutic Goods Administration (Australia), involve centralized, risk-based regulatory systems analogous to the centralized authority of the FDA. However, other regulatory contexts, such as the EU Notified Bodies, operate within decentralized frameworks where conformity assessment relies on harmonized standards (e.g., ISO 13485) and CE marking is determined through a client-service model.While structures may vary, pre- and postmarket review activities are similar across regulatory bodies. Review and enforcement components include premarket technical documentation review, postmarket surveillance, and compliance audits. These components operate within an interconnected regulatory framework that facilitates continuous oversight of medical devices. Premarket review processes inform postmarket surveillance strategies and postmarket data feeds into compliance and future regulatory decision making. The emphasis on, and significance of, each component continually evolves according to strategic priorities and local government oversight. For example, the FDA reorganized integrated reviews, surveillance, compliance, and quality into a single office in 2019 to create a regulatory framework that allows comprehensive oversight of medical devices across the product life cycle – from initial design and development to real-world use. This integration strengthened communication between pre- and postmarket regulatory activities and improved decision making.17 Similarly, the increased reliance on premarket clinical evidence and changes in device classifications under the EU MDR4 and EU IVDR5 have resulted in a more robust premarket review than under the previous EU Medical Device Directive (MDD).18,19 These examples illustrate why acquiring experience across the TPLC is key to career progression.
IHAs take various approaches to ensure that devices are safe, operate as intended, and deliver the anticipated patient benefit. Laws, regulations, local guidance, and recognized/harmonized standards determine how to introduce and maintain devices in a regulated market. For example, while FDA reviewers focus on direct determinations of safety and effectiveness or substantial equivalence for Class III and II devices, notified body experts evaluate compliance with General Safety and Performance Requirements (GSPRs) and ensure alignment with harmonized standards. For former FDA employees looking to transition into IHA roles, learning about the local technical documentation structure, national interpretation of standards (if applicable), International Medical Device Regulators Forum (IMDRF) documents that describe global harmonization efforts, and the Medical Device Single Audit Program (MDSAP) is critical.
Understanding country-specific regulatory structures, technical documentation requirements, and regional interpretations of standards is essential for professionals considering a transition into an IHA or notified body role. Resources such as IMDRF guidelines and the MDSAP can provide insight into global harmonization efforts. That said, the experiences and expectations of regulatory professionals may vary significantly depending on the organization, region, and regulatory framework in which they operate. Professionals seeking to transition to an IHA or notified body should seek appropriate mentorship, training programs, and professional networks to gain a deeper understanding of the challenges and opportunities of the specific role.
Other/nonprofits
Depending on their education and FDA experience, those who leave the FDA may find employment at organizations with smaller regulatory teams or missions other than product development, approval, and postmarket compliance. This section will explore these types of organizations and the opportunities for each type.- Healthcare delivery organizations (HDOs) deliver care to patients. HDOs may employ physician innovators or have centers that seek to develop novel technologies or improve existing medical technologies. HDOs routinely act as sites for clinical studies of investigational medical products and may house their IRBs. Regardless of whether the HDO is developing products or administering clinical studies, regulatory, quality, and clinical professionals are needed to help the HDO comply with applicable regulatory requirements.
- Trade associations represent and advocate for member companies and address challenges through policy advocacy work in a noncompetitive space. Several trade associations represent manufacturers of medical products, such as the Advanced Medical Technology Association (AdvaMed) and Medical Device Manufacturers Association (MDMA). These trade associations have historically welcomed former FDA officials, who can take on broader policy matters affecting member companies because of their unique insight gained through agency review or policymaking experience.
- Standards development organizations (SDOs) or voluntary consensus standards bodies are domestic or international organizations that plan, develop, establish, or coordinate voluntary consensus standards using agreed-upon procedures.20 US government policies have encouraged the development of consensus standards instead of government-developed standards (e.g., National Institute of Standards and Technology, Occupational Safety and Health Administration). This has led to increased opportunities at SDOs like the American National Standards Institute (ANSI), the Association for the Advancement of Medical Instrumentation (AAMI), ASTM International (ASTM, formerly the American Society for Testing and Materials), and others. SDOs may employ professionals with standardization experience to manage programs, projects, and engagement with external stakeholders.
- Public-private partnerships (PPPs) are collaborative agreements between the public (i.e., government) and the private (i.e., industry) sectors to collaborate on a topic of shared interest to both parties. Topics that may be addressed through PPPs include efforts to advance regulatory science, product development, manufacturing, and clinical trial infrastructure. Many PPPs operate through nonprofit organizations, like the Medical Device Innovation Consortium, the Foundation for the National Institutes of Health, and America Makes. PPPs may provide a structured forum for the FDA and industry to collaborate on complex problems before formal regulations or policies are proposed through rulemaking or the guidance process. Therefore, PPPs often leverage the expertise of former federal agency staff because of their requisite expertise, understanding of the problem, and ability to propose creative solutions that achieve shared goals.21,22
- Nonprofits include several types of organizations, such as foundations and advocacy organizations. For example, the Gates Foundation addresses poverty, disease, and inequity worldwide through grants and strategic investments. These foundations require significant expertise, including technical knowledge of scientific topics and regulatory and quality expertise, to be successful. Nonprofits also include patient advocacy organizations (PAOs), mission-driven nonprofit groups that seek to help people and their families affected by specific medical conditions.23 Stemming from efforts at the Center for Devices and Radiological Health (CDRH) and other FDA centers to increase patient engagement in the product development process, some PAOs have begun to hire beyond traditional health policy staff and have expanded their staffing to support regulatory science efforts with the FDA.
- Academic institutions encourage researchers to translate new ideas into commercially viable technologies via technology transfer and licensing offices. These opportunities are often catalyzed through formal programs that aim to equip and develop aspiring innovators with the skills necessary to launch a successful product onto the market. In many academic research institutions, instructors, mentors, administrators, and others help facilitate research follow-up efforts. In addition, some universities have a regulatory and quality group that, often in coordination with the licensing office, provides training and support through the product development and launch stages. These regulatory groups may participate in writing presubmissions and other submissions to the FDA.
- Third-party review organizations are permitted by the FDA to review certain low- to moderate-risk medical device submissions under the FDA 510(k) Third Party Review Program.24 These organizations sometimes hire former FDA. While a broad range of devices are eligible for third-party reviews, the eligible devices may not be as technically complex as higher-risk devices, with less specialized expertise required to understand their operation. The validation methods may also be better developed so that validation requires less in-depth analysis. Candidates for third-party review roles may, therefore, benefit from emphasizing the breadth and versatility of their experience, as opposed to their deep technical expertise on a single specific device.
- Other federal government agencies may be similar to the FDA regarding chain-of-command and operational structures and are another option for former FDA personnel. When transferring from one federal agency to another, the professional’s years of service are typically preserved. Knowledge of regulations, precedents, standards, and government databases can translate to other government agencies effectively. The application process and timelines for hiring and interviewing are similar to the processes at the FDA. While other government agencies have different mandates and scopes in their responsibilities to serve the public, the overall framework and activities may be familiar. Understanding the agency’s structure, mandates, and typical roles will be helpful for regulatory professionals considering transitioning to another agency.
Seniority/leveling
Like the FDA’s CDRH, many healthcare organizations use a dual-career ladder framework to support career growth for technical experts (individual contributors) and those who seek supervisory roles. However, job titles and reporting structures vary depending on the size and needs of the organization. The scope of regulatory affairs roles in small organizations tends to look different than that of large organizations, with regulatory professionals often wearing multiple hats in start-ups and small firms and supporting various activities, from clinical evaluation reports to quality system audits.Despite differences in structure and scope, most organizations provide an environment for employees to progress in their careers by advancing as subject matter experts or moving into progressively higher managerial roles with broader organizational responsibilities. While job titles and organizational structures vary between small organizations and MNCs, four distinct career levels emerge: entry-level specialists, mid-career strategists, senior technical leaders, and executive decision makers.25 As shown in Table 1, these tiers typically consist of technical and managerial progression tracks, offering comparable growth opportunities that scale with domain expertise in pay grade, influence, and recognition.
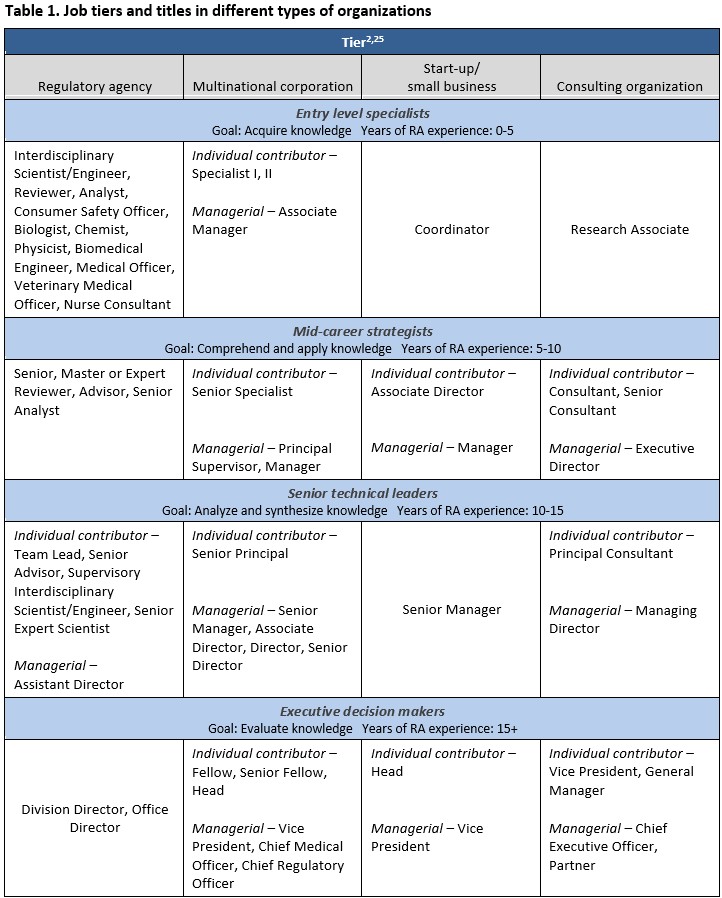
Former regulators transitioning into industry roles will find that corporate structures emphasize cross-functional collaboration, risk management, and alignment of regulatory strategy with business objectives. The organizational structure and culture may differ significantly between companies and parts of a single company. At smaller companies, regulatory activities may be grouped under clinical, quality, or other functional units. Regulatory professionals may report directly to the CEO or through another department, such as legal, but will typically be independent of marketing or sales departments. In larger organizations, first-line managers may supervise a few entry-level regulatory specialists who generally focus on tactical execution, including conducting preliminary regulatory assessments and maintaining technical documentation. As expertise develops, these managers tend to supervise more specialists across various related product lines and engage regulatory authorities in introducing and maintaining products in regulated geographies. In more senior-level roles, supervisory responsibility can expand to overseeing multiple regional regulatory teams, with senior-level technical experts working with cross-functional partners to develop clinical evaluations and global regulatory strategies. Finally, executive leadership roles shape organizational structure and governance, guide M&A activity, and set strategic priorities across the function.
The entry point into an organization will vary based on prior experience, domain expertise, and organizational needs. Table 1 provides examples of professional titles, although they will differ across organizations and cannot be directly compared with the government’s GS-level system. For example, not all organizations have a Vice President of Regulatory or a CRO.
Individual contributor and management pathways tend to diverge mid-career. These pathways often require distinct skill sets and experience. For example, specific management roles may require at least five years of management experience – including overseeing budgets and profit-and-loss sheets for a particular division or group – and demonstrated year-on-year growth. Most medical technology companies also look for managers with technical expertise, as they will need to lead cross-cutting teams dependent on the technical success of products. However, management positions vary across organizations based on the size of the company and the needs required at each stage. Some management roles, especially at smaller companies, may involve managing quality or clinical functions in addition to regulatory functions.
Section 2 – Job hunting
Unfortunately, job hunting is rarely a simple input/output exercise where an individual applies for a job and receives a response. In many instances, a company does not respond to the initial application (or does not do so promptly) or abruptly stops communicating during the interview process (e.g., after several interviews, the company stops providing updates). Amidst these frustrations, it can help to think of job hunting as a way to explore new opportunities and determine professional and personal values. For example, during an interview for a remote position, a company may disclose that most of the team is based on the West Coast, which may pose challenges for candidates based on the East Coast if, for example, the company emphasizes the need for frequent in-person interaction, or if the differences in time zones are not feasible to deal with daily.Networking
Establishing and maintaining a network is important even when not actively searching for a job. However, it is never too late to start creating a robust network and making connections. Consider it an exercise supporting current and future job searches since a network can be a powerful tool to stay on top of opportunities. In any event, having a wide network is a way to learn more about individuals, their interests, their organizations, and current and future opportunities. It is also an excellent opportunity to showcase one’s skills and potential contributions to professional contacts.Regarding networking, one should be open-minded, brave, and creative. Reach out to individuals with whom one has had positive professional interactions or follow their social media to see if they have posted any job listings or whether representatives from their companies will be attending upcoming conferences. Be mindful of ethical and recusal obligations. One should always seek advice from the FDA’s Ethics and Integrity Office if any situation might warrant further guidance.
Attending local networking events, industry conferences, and/or alumni events can be valuable, especially when learning about local employers and opportunities. Professional societies like RAPS have local chapters and networking groups. Local, self-organized medical technology meetings or regulatory associations can be free or inexpensive. In addition, there is an FDA Alumni Association for former FDA employees to discuss career goals and current news about the agency. As of the publication date of this article, lifetime membership costs $500 for alumni or $400 for those still employed by the agency and can rapidly pay for itself in connections and referrals.
Identifying opportunities and applying for a position
Although companies may prefer regulatory professionals with product knowledge who can effectively advise on validation strategy – especially when dealing with technically complex products such as surgical robotics or medical imaging – candidates with device-specific experience can also be challenging to find, especially if a company requires them to live in a particular geographic location. Therefore, former FDA employees typically do not need to limit themselves to applying for roles in the specific device area that they worked in at the FDA. Specific regulatory roles focus more on knowledge of regulatory processes, policies, and procedures. Even in more technical roles, procedural knowledge can often be sufficient to perform the role while coming up to speed on specific technology.Opportunities can be found on sites such as LinkedIn or Indeed, through recruiters, company websites, or by word of mouth. Some openings, especially for high-level positions, are not publicly advertised and may source candidates through specialized executive recruiters. Getting an official referral from an employee inside a company and familiar with one’s work can help during the interview stage. Once hired, the referring employees will often receive a referral bonus. Hiring in industry generally moves more quickly than with the FDA. Once a company has identified a top candidate and the back-up candidates, it is in its interest to close the hiring process as soon as possible. Suggested start dates are typically very soon – often at the beginning of the next pay period. After interviewing with a company, be prepared to make a rapid decision on an offer. Offers are typically conditional on passing background checks, which can take several days to a few weeks. Unlike at the FDA, a lengthy process involving fingerprinting and obtaining an official public trust clearance is not generally required.
Networking can often be the best method to identify opportunities. If one wishes to learn more about a company or organization and there are no current conflicts of interest, consider contacting anyone in the network who can connect with an employee there. A personal introduction will likely lead to a fruitful connection and productive conversation. Furthermore, discussions where competence is demonstrated, and a genuine interest in a company is expressed can sometimes lead to the creation of a position specifically tailored to the individual.
Continuing education and certificate programs
Continuing education is an important consideration for regulatory affairs professionals, regardless of their role or employer. Staying current through professional development helps maintain competence as the regulatory landscape evolves with new legislation, guidance documents, and industry standards. Learning opportunities through formal courses, industry conferences, or free webinars can help sharpen analytical skills and expand knowledge of global regulatory bodies.Regulatory master’s degrees, graduate programs, and certificate courses such as the Regulatory Affairs Certification (RAC) offer several benefits for regulatory affairs professionals while not required for career advancement. Some hiring managers view additional credentials as helpful in demonstrating specialized knowledge and commitment to regulatory expertise, potentially enhancing credibility with colleagues, management, and regulatory authorities. For professionals seeking career mobility or transitioning between sectors (medical devices, pharmaceuticals, or biologics), a master’s degree or RAC credential can serve as a recognized validation of their expertise. The RAC exam is oriented toward industry professionals, and studying for it may help in identifying gaps between the knowledge acquired in typical FDA roles compared with industry roles. Study groups are also valuable in building an initial industry network.
That being said, many successful regulatory professionals build distinguished careers without obtaining additional degrees or certificates. Industry experience, specialized therapeutic knowledge, and demonstrated regulatory competence often carry more weight than formal credentials. For example, the perceived value of RAC certification varies significantly by organization. Some employers value the credential, while others prioritize applicable experience and demonstrated capabilities. Professionals should evaluate their specific career goals, target organizations, and industry sector before investing time and resources in pursuing additional degrees.
Drafting or updating a resume
Drafting a resume using the STAR method (Figure) will highlight the applicant’s responsibilities and have an impact on results for the reviewers. This method quantifies performance rather than simply listing activities, increasing the impact on the reviewers. Use succinct bullet points and be prepared to expand on them during a behavioral interview.Figure. Outline of the STAR method27

Created by Caroline Rhim
In addition, aligning the text on a resume to the job description can enhance search engine optimization, as companies sometimes use technology to cull the many applications received for a vacancy. Using keywords from the job description may distinguish one resume from another. A master resume outlining all one’s experiences can be pared down to the requirements of a specific job description. To pare down experience to fit a specific role, include a selected experience heading at the top of the resume.
It is also worth highlighting that resumes for government and industry positions look different.26 While government resumes can be five or more pages, industry resumes may be only one to two pages. Therefore, it may be easier to condense a government resume to create a tailored resume that is appropriate for an industry position.
When applying to an industry role, any federal language on resumes should be translated into business language. Consider how review work encompasses more than the technical review and involves other skills such as project management, distilling complex information from multiple subject matter experts, and presenting information to stakeholders using clear and concise language. The GS level is not typically relevant. Use active language (e.g., lead, develop, manage.) to describe roles and responsibilities.
Interviewing
Phone screenAfter reviewing a resume, a recruiter may reach out for a phone screen. Although jobs advertised in the regulatory space typically receive hundreds of applications, only a fraction of those applications meet the minimum qualifications of the job advertisement. For example, it is common for a device software company to request device experience but receive many applicants from the pharmaceutical sector. Phone screens are offered to applicants whose resumes match the criteria that a hiring manager has provided to human resources. Phone screens vary in format and duration and may be as simple as confirming the details of the applicant’s resume. In other instances, a recruiter or human resources member may set up a call to understand the applicant’s interest and availability in a position. If a job description includes a salary range, be prepared to address questions regarding compensation. Companies will not want to expend resources on a candidate whose posted salary range is not aligned with their expectations. Be thoughtful about responding to salary expectation questions and gauge whether there is flexibility.
Behavioral interviews
Initial interviews may be behaviorally based and are used to understand a candidate’s ability to think quickly and thoughtfully, provide relevant examples, and assess past behavior as a predictor of future results. When preparing for a behavioral interview, it may be helpful to consider the STAR method,27 as shown in the preceding Figure.
Conducting a quick review of a company’s website and press releases can go a long way in demonstrating interest and familiarity with the company’s mission statement and product line development during an interview. It also may identify concerns or potential issues for the business and allow a candidate to highlight how they can help.
Technical interviews
After passing a behavioral interview with a human resources manager or recruiter, candidates are typically invited for technical interviews with the hiring manager and team. However, the interview process is not always linear, and subsequent interviews with the hiring manager may be behavioral. A technical interview can be conducted one-on-one or as a panel. It may be one interview or a series of interviews. It is important to review the job description and be prepared to provide evidence of qualifications.
Some interviews may also require writing samples during the interview process. If interviews are scheduled for a half day or a nearly full day on site, time may be set aside for the candidate to provide a written assessment of a business scenario. Alternatively, a company may request a writing sample to evaluate the ability to communicate an assessment of the situation, including a thorough consideration of the risks and rewards of options and recommendations.
When applying for a manager level or higher position, there may be competition with industry regulatory professionals who came up the traditional corporate ladder. The hiring manager may also have a similar background. Former FDA employees should be aware of their unique value as well as their experiential or knowledge gaps. Be aware that the number of regulatory submissions a candidate has written is often viewed as a metric of expertise for industry regulatory roles. Companies are occasionally skeptical that FDA review experience will translate into submission authoring experience. Be prepared that hiring managers may be concerned that although a candidate has read a hundred applications, they have never written one. Remember that while an FDA reviewer or consulting scientist may not have direct experience preparing submissions, they may have seen a greater number and wider variety of submissions in a year than many industry professionals will participate in across their entire careers.
Case interviews
Case interviews are not typical for regulatory functions. However, they are common in management consulting interviews, where a candidate is asked to analyze and solve a business problem. They are meant to assess a candidate’s analytical and soft skills in a real-world scenario. Case interviews are conducted one-on-one and allow the interviewee to work with and ask questions of the interviewer. Many online resources can help a candidate prepare for case interviews.
Employees leaving the FDA must adhere to ethical guidelines and cooling-off periods to prevent conflicts of interest when transitioning to industry roles.28 For additional questions, consult the FDA’s Ethics and Integrity Office.
Negotiating salary and evaluating offers
In addition to base salary, compensation packages in the industry may include healthcare benefits, 401(k) retirement fund and matching, a profit-sharing plan or bonus structure, stock options or RSUs, vacation time/paid time off, relocation package, and a signing bonus.2 These packages vary widely depending on the size of an organization and the geographical area. Potential areas of negotiation include salary, bonus structure, and, in some cases, vacation time/paid time off, signing bonus, equity, and the relocation package. Some organizations do not stipulate paid time off and have an unlimited leave policy. Employees who depart these organizations will not be paid for unused vacation time. With a few exceptions, the industry does not offer pensions or the ability to retain employer-sponsored health insurance after retirement. Due to the lack of these federal benefits, and since employment is at will, industry salaries are typically higher than government salaries at comparable career levels.Candidates may be asked for salary expectations during screening interviews. Recruiters should share the salary expectations with the candidates during the interview, but if one is not provided, insisting that they provide one based on years of experience or expertise is not recommended. If the job description does not provide a range, resources such as the RAPS compensation report2 and websites such as Glassdoor should be used to review salary ranges for similar job titles and locations. In the beginning stages of salary discussions with HR, candidates should provide a range, rather than a single number, that is supported by these resources. Always negotiate the salary, even if it only yields additional paid time off. It is common for a candidate who has received multiple job offers to negotiate for a higher salary by citing the highest salary offered by another company. Candidates do not have to share which companies have made competing offers.
Another difference between industry and government benefits is around health insurance. Government employees have more health insurance choices than those in industry. There are typically one to three levels of healthcare plans to choose from in industry. However, all are generally from the same insurer and are usually negotiated by the company for all employees. Healthcare expenses vary widely, and it is important to consider potential out-of-pocket expenses within salary negotiations.
While not negotiable, it may be important to understand which insurance network the company offers to determine if specific specialists and physicians are covered. It is also reasonable to ask if dental and vision insurance is included in the company’s healthcare plan and what kind of insurance plans they offer (e.g., preferred provider organization, high deductible), as each has its own rules and out-of-pocket expenses. If high-deductible plans are offered, determine if the company contributes to a healthcare savings account on behalf of the employee. Candidates may also want to ask if the company offers the same percentage of insurance premium coverage for their family and partner or if the employee must fully pay the healthcare premiums.
Many organizations offer employer matching for 401(k) contributions (though start-ups usually do not). This amount can vary from fifty cents on the dollar to dollar for dollar up to a certain max contribution amount. When comparing offers, it is important to know whether an organization offers employer matching, the matching rate, and how long one will need to remain at the company for the employer matching to vest. A financial adviser can assist former government employees with what to do with their thrift savings plan (TSP) upon leaving federal service. TSPs can typically remain open but not allow new contributions unless the individual returns to federal service.29
Employers may offer a relocation package to employees moving to a new state or region, depending on the job market, skillset, and position level. This can vary from completely covering relocation expenses based on reimbursement of receipts to a maximum relocation expense amount. It is rare for companies to help employees sell houses.
A one-time signing bonus may also be offered to a candidate. Companies are often more willing to negotiate on signing bonuses, a one-time expense, than salaries, a recurring expense. However, it is recommended to focus on negotiating the base salary, as subsequent raises are typically calculated as a percentage of an employee’s salary. Securing a higher salary from the start and receiving raises based on that salary may help negotiate a better salary for future roles.
Discussion and summary
FDA experience provides a strong foundation for many careers in the life sciences sector, equipping professionals with critical insight into US regulatory processes and translational skills in cross-functional collaboration. This section summarizes key themes discussed throughout the article, outlining career paths, transferable competencies, and their relevance across industry roles.Working at the FDA allows employees to impact public health through historically stable employment, strong benefits, and a relatively well-defined career path. It is difficult to generalize about private sector employment, which encompasses many types of organizations. However, private sector employment comes with different goals and a distinct risk/reward profile compared with federal employment. FDA employees serve the public by developing and applying regulations, whereas private sector employees focus on advancing market access, commercializing medical products, and growing business. Historically, the federal government has offered certain protections that most private-sector employers do not have. In exchange, companies may offer higher earning potential and can sometimes offer faster career advancement in a more dynamic environment.
Table 2 compares key aspects of the FDA and industry roles. Competencies such as effective time management, multitasking under competing and changing priorities, and leadership in interdisciplinary settings are important across the private sector. FDA experience offers transferable skills acquired by concurrently managing multiple submissions, working and negotiating with different sponsors, and leading or providing expertise to cross-functional teams. Former FDA employees, including reviewers and research scientists, bring a unique perspective to the industry through their understanding of how the agency thinks, allowing them to interpret subtleties in regulatory language fluently translate the FDA’s regulatory requirements into actionable advice and strategy. In-depth experience with the data requirements for marketing authorization enables former FDA employees to bridge the gap between regulatory strategy and practical product innovation.
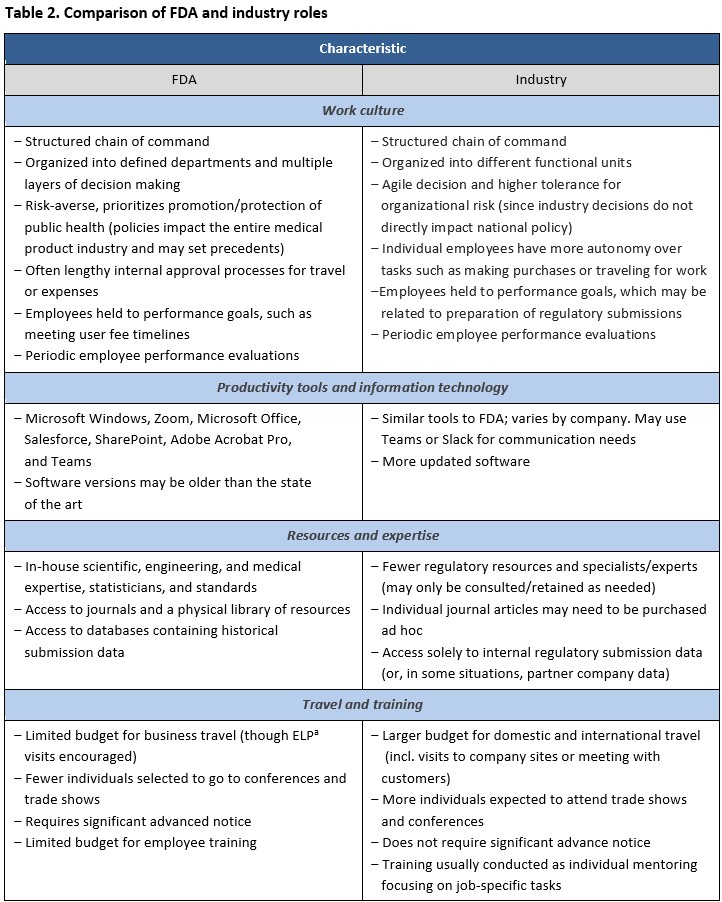
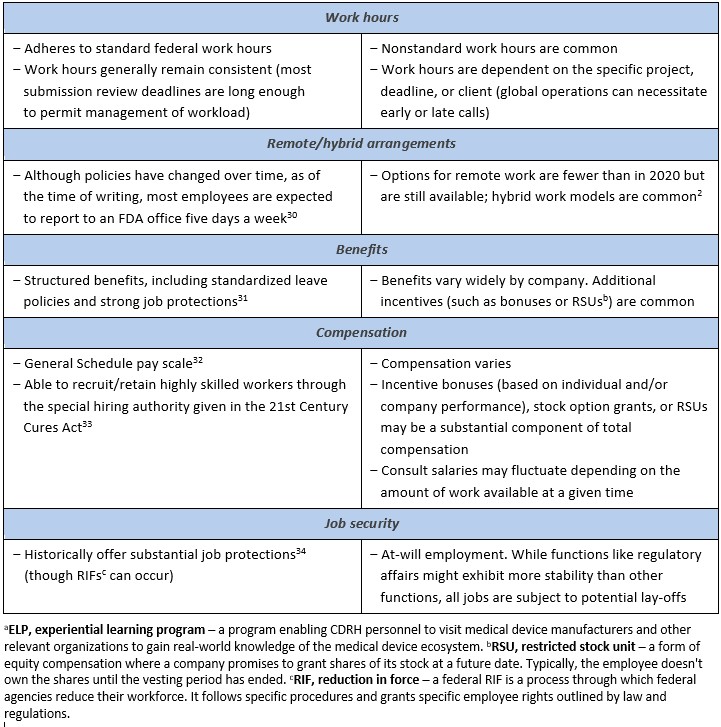
Former FDA employees should consider complementing their knowledge with international regulatory knowledge to better understand the evolving global regulatory landscape where regulations are increasingly interconnected across jurisdictions. A global outlook enables professionals to anticipate and adapt to varying regulatory requirements. This agile mindset facilitates the development of a harmonized strategy for submissions across various regulatory health agencies and ultimately streamlines patient access to medical products.
Former FDA employees may also wish to supplement their regulatory expertise with business and financial acumen. Unlike structured, user-fee-mandated timelines (e.g., MDUFA), industry settings are characterized by frequently shifting deadlines, priorities, and targets. At the FDA, a reviewer has the authority to determine that a medical product should not receive marketing authorization, provided that the decision is the result of appropriate review procedures. In industry settings, the regulatory professional’s role differs significantly. When a critical business need arises for a new product or a feature, regulatory affairs professionals are not expected to state that the FDA will not accept the submission. Instead, when challenges to commercializing products arise, regulatory professionals are expected to identify and adopt creative, solution-oriented approaches to navigate those challenges. Such approaches may include reducing the scope of labeling claims, marketing different versions of the product in different countries, or other compromises that help retain a product’s marketing value while satisfying regulatory agencies. The most effective industry regulatory professionals serve as strategic partners, offering viable solutions for companies to navigate constraints rather than merely identifying limitations.
The perspective required as an industry regulatory professional differs significantly from that required for FDA reviewers. A CDRH reviewer’s role is to decide whether the evidence submitted supports a device’s safety and effectiveness. On the industry side, it is of prime importance to identify the least burdensome pathway to marketing authorization and achieve the maximum possible marketing value that the data can support.
Especially within start-ups, where a company’s survival can hinge on rapid market entry, the ability to efficiently resolve regulatory inquiries and avoid protracted, inconclusive FDA discussions (e.g., many rounds of Q-submissions) is critical. Former FDA employees can highlight their experience in negotiating and facilitating the resolution of such issues as part of the value they offer to companies.
Given the constellation of functions that intersect with regulatory affairs, including quality, R&D, preclinical, clinical, medical affairs, product management, marketing, and sales, former FDA employees should develop a cross-functional understanding of the industry. Familiarity with NPD and design controls can be helpful, as can understanding the company’s quality system in a global context.
Beyond serving as an entry point into traditional regulatory affairs or consulting roles, FDA experience is also a pathway into clinical development, medical affairs, and preclinical safety. Although each field demands the acquisition of new skill sets, the foundational regulatory principles acquired at the FDA are applicable across the life sciences sector. FDA experience is even valuable in areas such as marketing – often perceived as the diametrical opposite of regulatory affairs – where a shared knowledge base can lead to career success when applied with tailored skills.
Conclusion
FDA experience provides a strong foundation for many careers in the life sciences sector. It equips professionals with critical insight into US regulatory processes and translational skills in cross-functional collaboration. This article has outlined some of the possible career paths, the value of the competencies gained at the FDA, and their applicability across industry roles.To enhance their career prospects, former FDA professionals moving to the private sector may wish to expand their expertise to include global regulatory frameworks, business strategy, and quality and compliance, among other areas. Compared with the more structured environment of regulatory agencies, industry roles focus on product development and commercialization. They may require increased agility, innovation, and strategic decision making to expedite market access while ensuring compliance with regulations.
Beyond regulatory affairs, FDA experience is valuable in a variety of roles, including clinical development, medical affairs, marketing, quality, and preclinical safety. This reflects the integration of regulatory considerations into broader business functions. Professionals who can combine FDA experience with a nuanced understanding of the industry have the potential to play key roles in shaping industry practices, bringing products to market, and advancing public health outcomes.
Abbreviations
AAMI, Association for the Advancement of Medical Instrumentation; AdvaMed, Advanced Medical Technology Association; AI/ML, artificial intelligence/machine-learning; ANSI, American National Standards Institute; ASTM, ASTM International [formerly American Society for Testing and Materials]; CDRH, Center for Devices and Radiological Health; CE, European Conformity; CFR, Code of Federal Regulations; CRO, contract research organizations; EU IVDR, European Union In Vitro Diagnostic Regulation; EU MDR, European Union Medical Device Regulation; FDA, [US] Food and Drug Administration; FSCA, field safety corrective action; GS, General Schedule; GSPR, General Safety and Performance Requirement; HDO, healthcare delivery organization; IDE, investigational device exemption; IHA, international health authority; IMDRF, International Medical Device Regulators Forum; IRB, institutional review board; ISO, International Organization for Standardization; IT, information technology; LLC, limited liability corporation; LTF, letter to file; M&A, mergers and acquisitions; MDD, Medical Devices Directive; MDMA, Medical Device Manufacturers Association; MDR, Medical Device Report; MDSAP, Medical Device Single Audit Program; MDUFA, Medical Device User Fee Agreement; MNC, multinational corporations; NPD, new product development; OSHA, Occupational Safety and Hazard Administration; PAO, patient advocacy organization; PMA, premarket approval application; PPP, public-private partnership; QSR, Quality System Regulation; R&D, research & development; RA, regulatory affairs; RAC, Regulatory Affairs Certification; SDO, Standards Development Organization; SOP, standard operating procedure; STAR, situation, task, action, result; STEM, science, engineering, technology, and/or mathematics; TPLC, total product lifecycle; TSP, thrift savings plan; UDI, unique device identification; V&V, verification and validation.About the authors
Jay Vaishnav, PhD, RAC-Devices, FRAPS, is the director of regulatory at Canon Medical Informatics. Her expertise is in medical imaging and radiotherapy, software as a medical device, software in a medical device, and artificial intelligence-enabled medical devices. In addition to more than seven years at the FDA and CDRH, Vaishnav has two decades of experience in science and technology at start-ups, multinational corporations, the National Institute for Standards and Technology, and academia. She co-edited RAPS’ Fundamentals of Medical Device Regulations: A Global Perspective and From X-Rays to AI: Navigating US Regulations in Radiological Health. She holds a PhD in physics from Harvard University, is a former assistant professor of physics at Bucknell University and teaches at the University of California Santa Cruz Silicon Valley Extension. Vaishnav is a RAPS member and can be reached through LinkedIn
James Kleinedler, PhD, is the director of regulatory strategy at Medtronic. He is a former device reviewer at the FDA, where he led premarket review of Class II & III cardiovascular device applications and served as a biocompatibility focal point within CDRH. He holds a doctorate in toxicology from Louisiana State University, has written 10 peer-reviewed publications, and is an inventor on seven medical device patents. Kleinedler is also an adjunct professor at St. Cloud State University’s graduate program in Regulatory Affairs. He is a RAPS member and can be reached through LinkedIn
Véronique Li, MBA, is a medical device regulatory expert at Hyman, Phelps & McNamara (HPM). Her expertise includes product development and authorization, compliance, due diligence, and internal and government investigations. Before joining HPM, she worked at Abbott and PwC. She is a former FDA lead reviewer and policy adviser in CDRH and CBER. She is certified in quality management systems and has a Six Sigma Green Belt. Li earned a BSE in Biomedical Engineering from Case Western Reserve University and an MBA in Healthcare Management from Boston University. She is a former adjunct professor at George Washington University. She can be reached through LinkedIn
Kristy Katzenmeyer-Pleuss, PhD, is the president and founder of KP Medical Device Consulting (KPMDC) and a former FDA premarket medical device reviewer. Katzenmeyer-Pleuss has 13 years of experience in the regulated medical device industry, over 20 years in the broader biomaterials field, and is a subject matter expert in biocompatibility and reprocessing. Before starting KPMDC, Katzenmeyer-Pleuss worked as a regulatory and biological safety consultant at NAMSA, a global CRO. Katzenmeyer-Pleuss holds a PhD in Bioengineering from the University of Washington and a BS in Chemical Engineering from the University of Wisconsin-Madison. She is a RAPS member. She can be reached through LinkedIn
Joshua Silverstein, PhD, is the director of regulatory science and policy at Philips. At Philips, Josh focuses on North America's new and changing regulations and policies, engagement with trade associations, public-private partnerships, and regulator initiatives. Silverstein joined Philips from Labcorp, where he was the associate director of regulatory affairs and global regulatory lead for companion diagnostics programs. He served in several roles for over 12 years at the FDA and CDRH as a regulatory scientist, premarket reviewer, and policy advisor. Silverstein received his bachelor’s degree in chemical engineering and PhD in Bioengineering from the University of Maryland. He is a RAPS member and can be reached through LinkedIn
Caroline Rhim, PhD, is the founder and principal consultant at Evera Regulatory Advisors and has over 20 years in the medical technology and life sciences fields. Rhim was previously the global business lead for the medical device consulting team at NSF Health Sciences, overseeing regulatory, QMS, and auditing workstreams. She brings experience as a manager for the orthopedic (spine) device team at CDRH and as a lead scientific reviewer in orthopedic and cardiovascular device groups. Rhim received her PhD in Biomedical Engineering from Duke University and her bachelor’s and master’s of science in materials science and engineering from MIT. She is a RAPS member and can be reached through LinkedIn
Nicholas Werner is the director of regulatory affairs (portfolio compliance) for CooperVision, where his current expertise is in regulatory intelligence and advocacy. He is a former FDA medical device reviewer and postmarket analyst. Before his current role, he held positions at Stryker Instruments and numerous consulting agencies, working in TPLC regulatory and with companies of all sizes and maturity levels. He received his bachelor's degree in bioengineering from the University of Pittsburgh. Werner is a RAPS member and can be reached through LinkedIn
Laura Gilmour, MS, is the founder and principal consultant of LG Strategies. Gilmour has worked in medical device additive manufacturing since 2009 and the medical device industry since 2006. She has held various roles, including key adviser to the Veterans Health Administration’s advanced manufacturing point of care efforts, global medical business development manager at EOS, R&D engineer at several medical device companies, and a premarket reviewer at the FDA. While at the FDA, Gilmore was a founding member of the Additive Manufacturing Working Group. Gilmour received master’s and bachelor’s of science degrees in biomedical engineering from the University of Tennessee and the University of Pittsburgh, respectively. She is a RAPS member and can be reached through LinkedIn
Allison Komiyama, PhD, RAC-US, FRAPS, is vice president of medical technology innovations at RQM+, a global medical technology service provider. As a former FDA reviewer and current regulatory consultant focused on FDA submissions, she has been involved in hundreds of presubmissions and premarket submissions for various medical device companies. She frequently presents at conferences, is an adviser at numerous technology incubators, and teaches students worldwide about regulatory submissions as a faculty member at San Diego State University Global Campus. Komiyama is a RAPS member, received the RAC in 2014, and became a RAPS Fellow in 2024. She can be reached through LinkedIn
Acknowledgment The authors thank Katherine Kim, MPH, RAC-Devices, for her support.
Disclaimer The views and opinions expressed here are those of the individual authors and do not necessarily reflect the views of any organization or entity with which they are currently or have previously been affiliated.
Citation Vaishnav J, et al. Life after the FDA: Career paths for former regulators. Regulatory Focus. Published online 25 March 2025. https://www.raps.org/News-and-Articles/News-Articles/2025/3/Life-after-the-FDA-Career-paths-for-former-regulat
References
- Medicare Payment Advisory Commission. Report to the Congress: Medicare and the health care delivery system – chapter 7: An overview of the medical device industry. Dated 2017. Accessed 28 February 2025. https://www.medpac.gov/wp-content/uploads/import_data/scrape_files/docs/default-source/reports/jun17_ch7.pdf
- Regulatory Affairs Professionals Society. Global compensation and scope of practice report for the regulatory profession. Dated 2024. Accessed 28 February 2025. https://www.raps.org/resources/scope-of-practice-survey
- 21 CFR Part 820, Quality system regulation. Published 1 April 2023. Accessed 28 February 2025. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820
- Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices. Published 5 May 2017. Accessed 28 February 2025. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745
- Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices. Published 5 May 2017. Accessed 28 February 2025. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R074
- ISO 13485:2016, Medical devices—Quality management systems—Requirements for regulatory purposes. International Organization for Standardization. Published 2016. Accessed 28 February 2025. https://www.iso.org/standard/59752.html
- ISO 14971:2019, Medical devices—Application of risk management to medical devices. International Organization for Standardization. Published 2019. Accessed 28 February 2025. https://www.iso.org/standard/72704.html
- IEC 62304:2006, Medical device software—Software life cycle processes. International Organization for Standardization. Published 2006. Accessed 28 February 2025. https://www.iso.org/standard/38421.html
- 21 CFR Part 50, Protection of human subjects. Last amended 25 February 2025. Accessed 28 February 2025. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-50
- 21 CFR Part 54, Financial disclosure by clinical investigators. Last amended 25 February 2025. Accessed 28 February 2025. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-54
- 21 CFR Part 56, Institutional review boards. Last amended 25 February 2025. Accessed 28 February 2025. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-56
- 21 CFR Part 58, Good laboratory practice for nonclinical laboratory studies. Last amended 25 February 2025. Accessed 28 February 2025. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-58
- 21 CFR Part 809, In vitro diagnostic products for human use. Last amended 25 February 2025. Accessed 28 February 2025. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-809
- 21 CFR Part 812, Investigational device exemptions. Last amended 25 February 2025. Accessed 28 February 2025. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-812
- No author] Small and midsize business (SMB). Gartner website. Not dated. Accessed 28 February 2025. https://www.gartner.com/en/information-technology/glossary/smbs-small-and-midsize-businesses
- Small Business Administration Office of Advocacy. Frequently asked questions about small business. Dated March 2023. Accessed 28 February 2025. https://advocacy.sba.gov/wp-content/uploads/2023/03/Frequently-Asked-Questions-About-Small-Business-March-2023-508c.pdf
- Food and Drug Administration. Total product life cycle of medical devices. Content current as of 6 September 2023. Accessed 28 February 2025. https://www.fda.gov/about-fda/cdrh-transparency/total-product-life-cycle-medical-devices
- Council Directive 93/42/EEC of 14 June 1993 concerning medical devices. End of validity 25 January 2021. Accessed 28 February 2025. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A31993L0042
- Dombrink I, et al. Critical implications of IVDR for innovation in diagnostics: Input from the BioMed Alliance Diagnostics Task Force. Published online 2022. Accessed 28 February 2025. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9126521/
- Office of Management and Budget. Circular A-119: Federal participation in the development and use of voluntary consensus standards and in conformity assessment activities. Dated 10 February 1998. Accessed 28 February 2025. https://www.whitehouse.gov/wp-content/uploads/2017/11/Circular-119-1.pdf
- Food and Drug Administration. Public-private partnerships. Content current as of 7 June 2023. Accessed 28 February 2025. https://www.fda.gov/emergency-preparedness-and-response/innovative-technologies/public-private-partnerships
- Food and Drug Administration. Case for quality. Content current as of 29 July 2020. Accessed 28 February 2025. https://www.fda.gov/medical-devices/quality-and-compliance-medical-devices/case-quality
- Rose SL. Patient advocacy organizations: Institutional conflicts of interest, trust, and trustworthiness. J Law Med Ethics. Published online 1 January 2021. Accessed 28 February 2025. https://doi.org/10.1111/jlme.12078.
- Food and Drug Administration. 510(k) third party review program. Content current as of 20 November 2024. Accessed 28 February 2025. https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/510k-third-party-review-program
- Regulatory Affairs Professionals Society. Regulatory Competency Framework. Dated 2021. Accessed 28 February 2025. https://www.raps.org/resources/regulatory-competency-framework
- Clabaugh J. How recently fired federal workers should reenter the job market. WTOP News. Published 224 February 2025. Accessed 28 February 2025. https://wtop.com/business-finance/2025/02/fired-feds-have-skills-its-a-matter-of-marketing-them/
- Office of Personnel Management. Structured Interviews: The STAR Method. Not dated. Accessed 28 February 2025. https://www.opm.gov/frequently-asked-questions/assessment-policy-faq/structured-interviews/
- Food and Drug Administration. Ethics. Content current as of 12 June 2019. Accessed 28 February 2025. https://www.fda.gov/about-fda/jobs-and-training-fda/ethics
- Federal Retirement Thrift Investment Board. TSP Fact Sheet: Information for TSP participants leaving federal employment. Dated February 2025. Accessed 28 February 2025. https://www.tsp.gov/publications/tspfs29.pdf
- Al-Faruque F. Federal workforce cuts, Trump buyout offer delayed, and FDA's RTO. Regulatory Affairs Professionals Society. Published 7 February 2025. Accessed 28 February 2025. https://www.raps.org/news-and-articles/news-articles/2025/2/this-week-federal-workforce-cuts%2C-trump-buyout-off
- Office of Personnel Management. Federal Employee Compensation Package. Not dated. Accessed 28 February 2025. https://www.opm.gov/policy-data-oversight/pay-leave/pay-administration/fact-sheets/federal-employee-compensation-package/
- Office of Personnel Management. Salaries & wages. Not dated. Accessed 28 February 2025. https://www.opm.gov/policy-data-oversight/pay-leave/salaries-wages/
- 21st Century Cures Act, HR 6. 114th cong. Dated 13 December 2016. Accessed 28 February 2025. https://www.congress.gov/114/plaws/publ255/PLAW-114publ255.pdf
- Government Accountability Office. Reduction in force can sometimes be more costly to agencies than attrition and furlough. Dated 24 July 1985. Accessed 28 February 2025 https://www.gao.gov/assets/pemd-85-6.pdf
Additional resources
- DiMichele L. The PhD scientist’s pathway into regulatory affairs. Regulatory Affairs Professionals Society. Published 16 April 2021. Accessed 18 February 2025. https://www.raps.org/news-and-articles/news-articles/2021/4/regulatory-career-development-the-phd-scientists-p
- Ekuta J, et all. Unlocking opportunities for entry-level positions in regulatory affairs. Regulatory Affairs Professionals Society. Published 27 August 2024. Accessed 18 February 2025. https://www.raps.org/news-and-articles/news-articles/2024/8/unlocking-opportunities-for-entry-level-positions
- McDermott O. Value of an RA qualification in continuous professional development. Regulatory Affairs Professionals Society. Published 26 April 2021. Accessed 18 February 2025. https://www.raps.org/news-and-articles/news-articles/2021/4/value-of-an-ra-qualification-in-continuous-profess
- Damrau N. The RAC examination: 12 tips for success. Regulatory Affairs Professionals Society. Published 5 June 2023. Accessed 18 February 2025. https://www.raps.org/news-and-articles/news-articles/2023/6/the-rac-examination-12-tips-for-success
- McDole-Russell M. Seven attributes for success as a regulatory leader. Regulatory Affairs Professionals Society. Published 8 December 2023. Accessed 18 February 2025. https://www.raps.org/news-and-articles/news-articles/2023/12/seven-attributes-for-success-as-a-regulatory-leade
- Hsieh I. A guide to finding the right mentor for regulatory affairs professionals. Regulatory Affairs Professionals Society. Published 29 July 2022. Accessed 18 February 2025. https://www.raps.org/news-and-articles/news-articles/2022/7/a-guide-to-finding-the-right-mentor-for-regulatory
- Rabaza R. 10 lessons learned during my first year in advertising and promotion regulatory affairs. Regulatory Affairs Professionals Society. Published 23 December 2020. Accessed 18 February 2025. https://www.raps.org/news-and-articles/news-articles/2020/12/10-lessons-learned-during-my-first-year-in-ad-prom
- FDA Alumni Association. Resources for FDA staff. Not dated. Accessed 18 February 2025. https://www.fdaaa.org/resources-for-fda-staff/
